 2020, Vol. 55
2020, Vol. 55



病毒感染一直持续地威胁着人类的生命健康。如今, 虽然已经成功开发出针对某些病原体[如人免疫缺陷病毒(human immunodeficiency virus, HIV)和丙型肝炎病毒(hepatitis C virus, HCV)]的有效抗病毒治疗方法, 但是对大多数病毒而言, 现在仍缺乏相应的抗病毒药物或疫苗[1]。有数百种病毒可以引起人类疾病, 包括HIV、HCV、乙型肝炎病毒(hepatitisB virus, HBV)、流感病毒(influenzavirus, Ⅳ)、日本脑炎病毒(Japanese encephalitis virus, JEV)、单纯疱疹病毒(herpes simplex virus, HSV)、人乳头瘤病毒(human papillomavirus, HPV)等[2]。尤其是在发展中国家, 已有的和新出现的病毒感染疾病日益严重[3], 这些病毒包括:登革热病毒(dengue virus, DENV)、黄热病病毒(yellow fever virus, YFV)、埃博拉病毒(Ebola virus, EBOV)、寨卡病毒(Zikavirus, ZIKV)、基孔肯雅病毒(Chikungunya virus, CHIKV)和甲型流感病毒(influenza A virus, IAV)、严重急性呼吸道综合征冠状病毒(severeacute respiratory syndrome coronaviruses, SARS-CoV)、中东呼吸道综合征冠状病毒(Middle East respiratory syndrome coronaviruses, MERS-CoV)等。特别是新型冠状病毒(SARS-CoV-2)感染疫情的暴发, 对于人类健康和社会公共卫生安全产生了极大的威胁(http://www.nhc.gov.cn/)。除了对症治疗外, 目前尚无确切有效的特异性治疗药物或疫苗, 凸显了研发抗病毒新药的迫切性。与此同时, 耐药株的快速出现也是迫切研发新一代抗病毒药物的另一个重要原因。
众所周知, 特异性的疫苗和小分子化学药物仍然是对抗病毒感染最强大的“武器”[4, 5]。与此同时病毒通过快速变异, 针对现有药物出现耐药性, 极大地削弱疫苗和抗病毒药物的作用[6, 7]。广谱抗病毒药物可以作用于多种病毒或同一种病毒的多种基因型, 相较于特异性抗病毒药物具有明显的优势[8-10]。
目前开发广谱抗病毒药物的策略主要有两种, 分别是干预病毒感染性和通过靶向病毒复制所必需的宿主因子来调节宿主细胞防御系统[11]。此外, 药物再定位策略作为开发广谱抗病毒药物的常用方法, 其可以绕过药物开发早期阶段所需要投入的时间与经费, 可以加速抗病毒药物的开发过程[12]。本文综述了广谱抗病毒药物的研究进展, 重点是调节宿主细胞防御系统的广谱抗病毒药物。
1 作用于病毒复制过程中特定病毒靶点目前主要通过干预病毒感染的必需步骤来控制病毒感染, 包括干预病毒吸附、融合、内吞、复制、装配、出芽等过程以及靶向病毒包膜的药物[11]。本部分主要介绍了作用于聚合酶和病毒包膜的广谱抗病毒药物。
1.1 作用于病毒聚合酶的广谱抗病毒药物病毒聚合酶是不同病毒家族中最保守的酶类, 在有显著的氨基酸序列差异的情况下, 它仍然保持着相同的整体结构和功能基序[13, 14]。病毒聚合酶是一个理想的抗病毒靶标, 已被一些成功的聚合酶抑制剂(例如阿昔洛韦、索非布韦)和核苷以及非核苷逆转录酶抑制剂所证实。因此, 聚合酶是开发广谱抗病毒药物的合适靶标, 作用于该靶标的广谱抗病毒药物可以同时阻断大多数含有聚合酶的病毒复制。
聚合酶作为广谱抗病毒药物靶标的代表性实例是致泻病毒的病毒聚合酶抑制剂的发现。图 1中列出了一些抑制杯状病毒或轮状病毒的RNA依赖的RNA聚合酶(RNA dependent RNA polymerase, RdRp)的核苷类似物(1~3)和一些具有抑制人腺病毒DNA聚合酶活性或阻断人腺病毒复制的核苷类似物(4~6)。其中代表性的化合物利巴韦林(1)对人诺如病毒(humannoroviruses, HNoV)具有体外抑制活性[15], 此外它还可以抑制HSV-1、HSV-2、人巨细胞病毒(humancytomegalovirus, HCMV)、IAV、乙型流感病毒(influenza B virus, IBV)、杯状病毒(calicivirus)、轮状病毒(rotavirus)等的复制[16]。法匹拉韦(2)能够抑制流感病毒RNA的合成, 而对人体DNA和RNA没有影响。各种研究表明, 除流感病毒外, 该药还对多种RNA病毒展现出良好的抗病毒作用, 如EBOV、沙粒病毒、布尼亚病毒、狂犬病毒等, 因此也被日本列入国家战略储备药物[17]。西多福韦(4)被激酶磷酸化后与病毒DNA聚合酶相互作用, 可终止病毒DNA链的延伸。核苷类似物通常被细胞酶类磷酸化为三磷酸活性形式, 并通过病毒聚合酶催化进入到新合成的核酸链中, 从而终止病毒核酸链的延长或者产生病毒诱变, 进而发挥抗病毒作用[18]。

|
Figure 1 The structures of compounds 1-7 |
2017年, Siegel等[19]发现瑞德西韦(7)可以抑制EBOV的复制, 其EC50值为86 nmol·L-1。Tchesnokov等[20]证实瑞德西韦通过抑制EBOV的RdRp来发挥抗病毒作用。此后进一步研究表明瑞德西韦可通过抑制病毒的RdRp对一系列的RNA病毒发挥作用, 如纤丝病毒科、副黏病毒科以及冠状病毒科[21-23]。值得注意的是, 最新研究发现, 瑞德西韦可以在低微摩尔浓度下有效地阻断SARS-CoV-2的感染(EC50 = 0.77 μmol·L-1, CC50 > 100 μmol·L-1, SI > 129.87)[24]。
1.2 作用于病毒包膜的广谱抗病毒药物据估计, 动物宿主中约有50万种未知的病毒[25]。与无包膜病毒相比, 包膜病毒更容易在不同物种之间传播[26, 27]。细胞能够通过细胞质内膜迅速修复细胞膜损伤, 但病毒颗粒无法修复受损的病毒包膜[28]。由于包膜脂质成分来自于宿主细胞, 与靶向经典病毒成分的那些靶标相比, 病毒更难对靶向病毒包膜的抗病毒药物产生耐药性。因此, 对于高突变率的病毒而言, 病毒包膜也是非常有前景的靶标[1]。此外, 靶向病毒包膜可以不必考虑透膜性的可能障碍。在这种情况下, 寻找针对病毒包膜的抗病毒分子并将其开发为广谱抗病毒药物具有广阔的前景[8]。
Chen等[29]发现分泌型磷脂酶A2 (secreted phospholipase A2, sPLA2)可以作为广谱抗病毒药物, 其作用机制为靶向来自内质网膜的病毒包膜脂双层。他们发现分泌型磷脂酶A2的CM-Ⅱ异构体(CM-Ⅱ-sPLA2)能抑制HCV、DENV和JEV的复制, 其EC50值分别为0.036、0.31和1.34 ng·mL-1。这些病毒的共同特点是通过内质网膜出芽; 相反, CM-Ⅱ-sPLA2对通过细胞质膜或反式高尔基体网络(trans-Golgi network, TGN)出芽的病毒的EC50值则大于10 000 ng·mL-1。此外, CM-Ⅱ-sPLA2的CC50和半数细胞溶血性浓度(HC50)都大于10 000 ng·mL-1, 这意味着CM-Ⅱ-sPLA2没有显著损害细胞质膜。这些结果表明, CM-Ⅱ-sPLA2及其衍生物是开发靶向来自内质网膜的病毒包膜脂质双分子层的广谱抗病毒药物的优质先导化合物。
2 作用于宿主蛋白的广谱抗病毒药物病毒复制依赖于宿主细胞, 需要宿主细胞提供必要的酶和底物。抑制病毒复制过程中必要的宿主蛋白(如酶类和受体)的功能可以达到抑制病毒复制的目的。本部分重点介绍了溶酶体、人腺苷三磷酸酶(RNA helicase X-linked DEAD-box polypeptide 3, DDX3X)、宿主激酶、亲环素A、Toll样受体、二氢乳清酸脱氢酶(dihydroorotate dehydrogenase, DHODH)作为广谱抗病毒药物靶标的研究进展。
2.1 作用于宿主溶酶体的广谱抗病毒药物为了进入宿主细胞, 病毒通常与宿主细胞表面的特定分子相结合, 诱导宿主细胞通过内体/溶酶体途径将其内吞[30]。为了感染宿主细胞, 病毒同时需要内体/溶酶体的酸性环境和酸性pH依赖性的内体蛋白酶(acidic pH dependent endosomal proteases)催化裂解糖蛋白[31]。没有内体酸化与糖蛋白裂解过程, 病毒的复制与感染就无法进行。
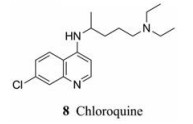
氯喹二磷酸盐是被FDA批准的抗疟药, 被证明是最成功的抗疟药, 因其具有较好的抗寄生虫活性与较低的毒性[32]。氯喹(8)作为一种典型的阳离子两亲性药物, 在生理条件下, 氨基部分主要以非质子化形式存在, 其可以自发且快速地穿过细胞膜或细胞器膜, 进入到酸性的内体/溶酶体或高尔基体内。随着酸性细胞器内的氯喹浓度升高, 其可以结合两个质子, 质子化后的氯喹不易穿过细胞器膜, 致使细胞器中的pH值升高, 其生理学与形态学性质均会改变, 包括病毒蛋白水解过程和翻译后修饰过程所需要的酶失去活性[33]。与此同时, 氯喹可以抑制INF-γ和TNF-α等细胞因子的产生, 还可以下调TNF-α受体的表达, 进而发挥其抗病毒作用[34]。
已有研究证明, 氯喹可在体外抑制多种病毒的复制, 包括HIV (氯喹凝胶表现出剂量依赖性的体外活性, 平均EC50值为从23~0.4 mg gel·mL-1)[35]、SARS-CoV (EC50 = 4.1 μmol·L-1, CC50 > 128 μmol·L-1, SI > 31)[36]、MERS-CoV (EC50 = 3 μmol·L-1, CC50 = 58.1 μmol·L-1, SI = 19.4)[36]、DENV (在非毒性浓度时剂量依赖性地抑制DENV复制)[37]、EBOV (EC50 = 16 μmol·L-1, CC50 > 50 μmol·L-1, SI > 3.1)[38]和ZIKV (EC50 = 9.82 μmol·L-1, CC50 = 134.54 μmol·L-1, SI = 13.70)[39]等。值得注意的是, 氯喹也可以抑制SARS-CoV-2 (EC50 = 1.13 μmol·L-1, CC50 > 100 μmol·L-1, SI > 88.50)[24]。
|
人腺苷三磷酸酶或者DDX3X是一种RNA解旋酶, 其属于从细菌到人类体内都保守的DEAD盒解旋酶家族[40, 41]。依据保守序列之间的相似性, RNA解旋酶被划分为超家族1~6。与其他DEAD盒蛋白一样, DDX3X几乎参与到RNA代谢的所有方面[42, 43]。DDX3X是一种核质穿梭蛋白, 是调控转录、翻译、RNA剪接、RNA转运和RNA降解过程的参与者之一[44]。除此以外, DDX3X在细胞周期和细胞凋亡的调节中也发挥一定的作用[45]。DDX3X还通过与Ⅰ型干扰素旁路途径的特定蛋白质相互作用来增强固有免疫系统的抗病毒能力[46]。因此, DDX3X是病毒复制所需的宿主因子和宿主抗病毒固有免疫应答的一个组分[47]。DDX3X在病毒复制和基因表达中的作用通常需要其ATP酶或者RNA解旋酶活性, 而其在固有免疫中的作用大多与其催化活性无关[48]。DDX3X在几种不同病毒的生命周期中发挥作用, 如HIV-1、HBV、HCV、WNV、DENV、IAV、沙粒病毒(arenaviruses)、疱疹病毒(herpesviruses)等, 对于其中许多病毒目前尚无可用的治疗方法。鉴于此, DDX3X被认为是有前景的抗病毒药物靶标。
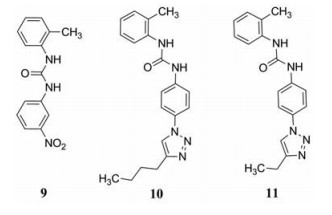
2012年, Radi等[49]设计并合成了第一个DDX3X小分子抑制剂, 特异性靶向其RNA结合位点。其中, 化合物9最具潜力, 具有对DDX3X的抗解旋酶活性(IC50 = 1 μmol·L-1)和对外周血单核细胞中HIV-1复制的抑制作用(EC50 = 10 μmol·L-1, CC50 = 100 μmol·L-1)。在此基础上, Brai等[8]利用基于结构的优化策略以及骨架跃迁策略对化合物9进行了优化, 发现了活性增强的化合物10和11, 其抑制DDX3X的IC50值分别为0.3和0.98 μmol·L-1。
|
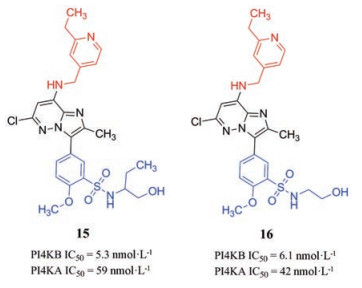
宿主激酶可以作为开发广谱抗病毒药物的靶标, 一个代表性的实例是PI4KB抑制剂的发现。阳性单链RNA病毒在一个称作“复制工厂”的特殊区域内复制[50]。这些膜衍生的区域富含磷脂酰肌醇4-磷酸(phosphatidylinositol-4-phosphate, PI4P)[51]。人类有4种合成PI4P的激酶, 即两种典型的脂质激酶: PI4KA和PI4KB, 以及两种Ⅱ型激酶: PI4K2A和PI4K2B[52, 53]。2017年, Mejdrova等[54]发现了可用于广谱抗病毒药物的新型高选择性磷脂酰肌醇4-激酶Ⅲβ (PI4KB)抑制剂。如图 2所示, 他们通过分子杂合策略将两种已知的PI4KB抑制剂12的蓝色部分和13的核心母环以及红色部分(化合物12选择性不高但活性较高, 其对PI4KB的IC50 = 19 nmol·L-1, 对PI4KA的IC50 = 1.1 μmol·L-1[55]; 化合物13选择性较高, 其对PI4KB的IC50 = 54 nmol·L-1, 对PI4KA的IC50大于100 μmol·L-1 [56])进行组合, 设计合成了一系列新型PI4KB抑制剂。经活性评价获得了许多具有优异抑制活性并保持对PI4KB选择性的化合物, 其中活性与选择性最好的是化合物14, 其对PI4KB的IC50为0.98 nmol·L-1, 对PI4KA的IC50为48 000 nmol·L-1。

|
Figure 2 The rational design of PI4KB inhibitors |
与其他PI4Ks抑制剂相比, 化合物14的抑酶活性达到亚纳摩尔水平, 是当时活性最高的PI4KB抑制剂, 而且其选择性较高, 相较于PI4KA的选择性指数SI值(selectivity index, SI)约为50 000。化合物14与PI4KB蛋白的分子对接结果(PDB:5FBL)中可以看到14的砜基氧原子、与咪唑并[1, 2-b]哒嗪环相连的仲胺基、酰胺的羰基分别与LYS-564的ε-氨基、VAL-613的羰基、ASN-615的α-氨基形成3个氢键, 有助于其与目标蛋白口袋的紧密结合, 进而发挥PI4KB抑制活性(图 3)。然而, 化合物14的抗病毒活性不高, 可能与其细胞透膜性差有关[54]。化合物15与16均具有纳摩尔水平的PI4KB抑制活性, 而且15的PI4KB抑制活性优于16, 但是在细胞实验中只有16表现出与体外一致的抗病毒活性。此外, 16对PI4KB亦具有选择性, 其对PI4KB和PI4KA的IC50分别为6.1 nmol·L-1和42 nmol·L-1 [54]。
|

|
Figure 3 The docking results of compound 14 and PI4KB protein complex (PDB: 5FBL) |
亲环素A (cyclophilin A, CyPA)是一种肽基-脯氨酰顺/反异构酶(peptidyl-prolyl cis/trans isomerases, PPIase)[57]。环孢菌素A (ciclosporin A, CsA)是FDA批准的药物, 用于器官移植时避免产生免疫排斥反应, 它与CyPA的结合可以抑制PPIase的活性, 从而发挥对HPV、VSV、HIV-1、HCV等的广谱抗病毒作用[58-63], 这是药物再定位策略应用于广谱抗病毒药物发现的一个实例。在保持CsA抗病毒活性的同时, 为了降低其免疫抑制活性, 已经开发一些新型的CsA衍生物, 例如alisporivir、NIM811和SCY-635, 其中alisporivir已在HCV患者中进行了Ⅱ期和Ⅲ期临床试验, 其安全性和疗效均表现良好[12]。
在此基础上, Ahmed-Belkacem等[57]运用基于片段及基于靶标的药物设计策略来发现新的非肽类小分子亲环素抑制剂。其中化合物5a在体外可以有效抑制CyPA、CyPB和CyPD的PPIase活性, 5a及其类似物对HCV、HIV和HCoV-229E复制有抑制作用(表 1)[57]。
| Table 1 EC50 (μmol·L-1) and CC50 (μmol·L-1) of the cyclophilin inhibitors against HCV, HIV and HCoV-229E, and cellular toxicities in vitro in their respective cellular models[57] |
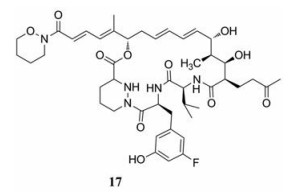
此外, 2015年, Hansson等[64]在sanglifehrin A的类药性优化过程中利用半合成与同源重组等生物工程相结合的方法合成了NVP018 (17)。NVP018具有体外抑制HBV、HCV和HIV-1的活性(表 2), 其EC50值分别为950、18和130 nmol·L-1。在小鼠、大鼠和犬体内, NVP018具有良好的口服生物利用度, 按照5 mg·kg-1的剂量口服给药后血药浓度保持在对HCV的EC90以上的时间为24~48 h。
|
| Table 2 EC50 (nmol·L-1) of cross genotype HCV replicon inhibition and IC50 (nmol·L-1) of HIV-1 and HBV inhibition[64] |
固有免疫系统通过模式识别受体(pattern-recognition receptors, PRRs)来识别病原体[65]。PRRs识别一组进化过程中高度保守的病原体结构, 即病原体相关分子模式(pathogen-associated molecular patterns, PAMPs), 从而诱导固有免疫反应的激活[66]。固有免疫的激活会导致被感染细胞和免疫细胞产生促炎症细胞因子和趋化因子, 其可以限制病毒复制并协调抗病毒适应性免疫[67]。PRRs有几个亚组, 其中Toll样受体(Toll-like receptors, TLRs)是哺乳动物中第一个被鉴定的也是研究得最为透彻的一个PRR[66]。TLRs的配体由于有诱导先天免疫和协调适应性免疫的能力而具有很大的抗病毒治疗潜力[68]。
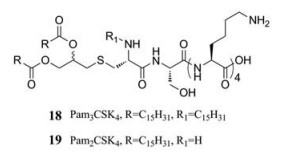
Du等[66]发现在HBV感染过程中TLR2信号通路对抑制病毒复制起着重要作用, TLR2激动剂有望成为治疗慢性HBV感染的新选择。化合物18、19是一些代表性TLR2激动剂的结构。此外, 在测试TLR配体作为治疗HIV感染中的免疫调节剂的试验中发现使用Poly (I:C) (聚肌苷酸-聚胞苷酸)或CpG-ODN (CpG寡脱氧核苷酸)分别刺激TLR3或TLR9是一种有效的暴露后治疗的方法[69, 70]。此外, Wang等[71]发现在恒河猴感染实验中, 先用TLR7和TLR9配体处理, 再用猴免疫缺陷病毒(simian immunodeficiency virus, SIV)进行感染, 也获得了类似的结果。这表明用TLR配体对逆转录病毒进行处理可以诱导保护性免疫, 进而发挥抗病毒作用[66]。
|
由嘌呤和嘧啶组成的细胞核苷酸在构成RNA和DNA中起重要作用。从头合成和补救合成是体内核酸合成的两种途径[72]。病毒复制严重依赖于宿主供应的核苷。因此, 参与核苷生物合成的宿主细胞内的酶类是开发抗病毒药物的另一个潜在靶标[73]。二氢乳清酸脱氢酶(dihydroorotate dehydrogenase, DHODH)是嘧啶从头生物合成途径中的限速酶[74]。DHODH位于线粒体内膜, 并在线粒体内膜上将二氢乳清酸转化为乳清酸[75]。然后, 多功能尿苷单磷酸合酶以乳清酸为底物催化合成尿苷单磷酸(uridine monophosphate, UMP), UMP是合成所有其他嘧啶核苷酸的必需前体之一。结果证实, 抑制DHODH活性可以抑制一系列病毒的复制, 包括EBOV、VSV、ZIKV等[76-79]。除了抑制病毒RNA合成外, DHODH抑制剂可以触发干扰素刺激基因(interferon-stimulating genes, ISGs)的表达, 这种活性可能也有助于这些化合物的抗病毒作用[80-82]。
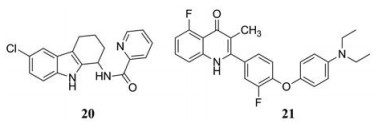
2018年, Luthra等[77]发现了抑制埃博拉病毒复制的小分子SW835 (20), 另一种DHODH抑制剂布喹那也可以抑制EBOV复制, 其EC50分别为9 nmol·L-1和0.15 μmol·L-1, CC50均大于50 μmol·L-1。除此以外, 20和布喹那还可以抑制水泡性口炎病毒(vesicularstomatitis virus, VSV) (EC50分别为0.4和0.5 μmol·L-1, CC50均大于50 μmol·L-1)和ZIKV (EC50分别为0.04和0.3 μmol·L-1, CC50均大于50 μmol·L-1)的复制[78]。
此外, 清华大学饶燏课题组发现了一种靶向DHODH的新型强效广谱抗病毒小分子RYL-634 (21)。它对各种致病性病毒表现出优异的广谱抑制活性, 包括HCV (EC50 = 4 nmol·L-1, CC50 > 2.5 μmol·L-1)、DENV (EC50 = 7 nmol·L-1, CC50 > 2.5 μmol·L-1)、ZIKV (EC50 = 20 nmol·L-1, CC50 > 2.5 μmol·L-1)、CHIKV (EC50 = 260 nmol·L-1, CC50 > 2.5 μmol·L-1)、肠道病毒71 (Enterovirus 71, EV71) (EC50 = 4 nmol·L-1, CC50 > 2.5 μmol·L-1)、HIV (EC50 = 13 nmol·L-1, CC50 > 1.25 μmol·L-1)、RSV (EC50 = 60 nmol·L-1, CC50 > 2.5 μmol·L-1)等[83]。
|
药物化学领域存在一些优势结构, 具有优势结构的分子往往具有某些特定的优异的药学性质。本部分总结了应用于广谱抗病毒药物开发的一些优势结构, 重点介绍了苯并咪唑衍生物、硝唑尼特、吩噁嗪类、绕丹宁和硫代巴比妥类以及一些天然产物作为广谱抗病毒药物的研究进展。
3.1 苯并咪唑类广谱抗病毒药物苯并咪唑是吲哚和嘌呤核的生物电子等排体。吲哚和嘌呤是天然核苷酸的结构单元, 存在于许多基本细胞成分和生物活性化合物中[84]。因此苯并咪唑类化合物很容易与生命系统的生物聚合物相互作用[85]。基于此, 苯并咪唑是抗病毒研究领域一类经典的优势结构。苯并咪唑衍生物作为广谱抗病毒药物的一个代表性实例是发现抗柯萨奇病毒B5 (coxsackievirus B5, CVB5)、抗RSV和抗Sb-1 (Poliovirus type-1, sabin strain, Sb-1)的新结构类型的小分子。
Tonelli等[86]设计并合成了一个由86个苯并咪唑衍生物组成的化合物库。测定了这些化合物对一组10种RNA和DNA病毒的抗病毒活性(表 3~5)。
| 表 3 EC50 (μmol·L-1) and CC50 (μmol·L-1) of 1-/2-phenylbenzimidazole derivatives[86] |
| Table 4 EC50 (μmol·L-1) and CC50 (μmol·L-1) of 2-benzylbenzimidazole derivatives[86] |
| Table 5 EC50 (μmol·L-1) and CC50 (μmol·L-1) of 1-/2-(non-aromatic substituted) benzimidazole derivatives[86] |
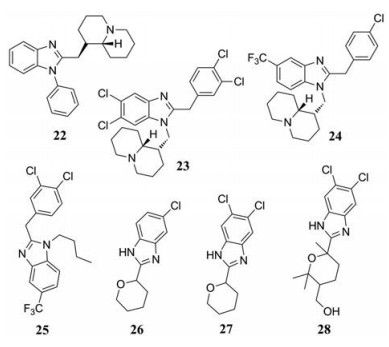
可以看出, 许多化合物对一种或多种病毒表现出不同程度的活性, 可作为新的苗头化合物。表 3和表 4的活性数据表明化合物22、23、24、25对CVB-5 (EC50值分别为17、9、13和12 μmol·L-1)和RSV (EC50值分别为7、12、15和9 μmol·L-1)均具有良好的活性。表 5数据表明, 通过对26、27、28 (EC50值分别为20、60和12 μmol·L-1)的2- (四氢吡喃-2-基)苯并咪唑骨架进行修饰, 可提高对Sb-1感染的抑制作用。
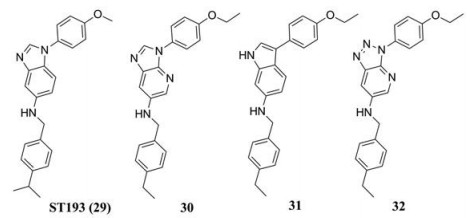
此外, Burgeson等[87]发现一系列基于苯并咪唑骨架的沙粒病毒抑制剂, 代表化合物为ST-193 (29)。结构优化集中在苯并咪唑骨架上, 包括引入各种取代基或者变动核心杂环, 最终发现了对拉沙病毒(Lassa virus pseudotypes, LASV)具有亚纳摩尔活性的化合物30, 其EC50值为0.16 nmol·L-1, 并且测定了30对包括JUNV、马秋波病毒(Machupovirus, MACV)、萨比亚病毒(Sabiá virus)和瓜纳里托病毒(Guanarito virus)的活性, 其EC50值分别为7.9、28、54和3.5 μmol·L-1。在此基础上, 他们利用生物电子等排原理对苯并咪唑环进行等排替换, 获得了化合物31和32, 其抑制LASV复制的EC50值分别为13和0.87 nmol·L-1。
|
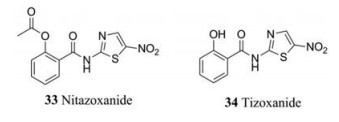
硝唑尼特(33, nitazoxanide, NTZ)是一种已被FDA批准上市的噻唑类化合物, 用于治疗由寄生虫、原虫和厌氧细菌引起的肠炎[88, 89]。NTZ不会影响IAV病毒吸附或侵入靶细胞, 不会抑制病毒蛋白表达, 然而它选择性地阻断病毒血凝素的成熟和细胞内转运[90]。NTZ口服吸收之后在血液中迅速脱乙酰化, 形成活性代谢物替唑尼特(34, tizoxanide, TIZ)[91]。TIZ对一系列厌氧菌具有抗菌活性[92]。研究表明NTZ的作用机制为阻断厌氧能量代谢所必需的丙酮酸-铁氧还蛋白氧化还原酶(pyruvate-ferredoxin oxidoreductase, PFOR)酶依赖性电子转移反应[92]。对结核分枝杆菌的研究表明, NTZ可作为解偶联剂破坏细胞膜电位和机体内pH稳态[93]。
|
Rossignol[92]发现TIZ可以抑制16种A型流感病毒株和1种B型流感病毒株的复制, EC50值范围为0.2~1.5 μg·mL-1, 发现TIZ还可以抑制副流感仙台病毒(parainfluenza Sendai virus) (EC50 = 0.1 μg·mL-1)、呼吸道合胞病毒A2 (respiratory syncytial virus A2) (EC50 = 0.3 μg·mL-1)和犬冠状病毒S-378 (canine coronavirus S-378) (EC50 = 1 μg·mL-1)的复制。另外, 根据Jasenosky等[94]最新的研究结果, NTZ广泛扩增细胞质RNA传感和Ⅰ型干扰素通路, 并有效地抑制EBOV和VSV的复制。当NTZ浓度为20 μmol·L-1时, 完全抑制EBOV增值; 当NTZ浓度为12 μmol·L-1时, VSV-Luc抑制率大于99%;而当NTZ浓度为18 μmol·L-1时, VSV-Luc抑制率大于99.9%。武汉病毒所及军事医学科学院研究表明, NTZ可以在低微摩尔浓度下抑制SARS-CoV-2 (EC50 = 2.12 μmol·L-1, CC50 > 35.53 μmol·L-1, SI > 16.76)[24]。
|
除此以外, 测定了TIZ的其他抗病毒活性。在使用猴肾MA104细胞的单周期病毒生长试验中, TIZ抑制猴SA11-G3P和人Wa-G1P的复制, 其EC50值分别为0.5和1 μg·mL-1; 而在多周期试验中, TIZ抑制轮状病毒和诺如病毒的复制, EC50为0.3 μg·mL-1[95, 96]。TIZ在2.2.15细胞中表现出对HBV复制的选择性抑制, 其EC50值为0.06 μg·mL-1, 并且对HCV基因型1a表现出抑制活性, EC50和EC90分别为0.09和0.38 μg·mL-1 [97]。同时, 在细胞培养试验中TIZ抑制黄病毒科的DENV-2、JEV和YFV的复制, 其EC50值为0.1、0.12和0.06 μg·mL-1 [98]。表 6列出了TIZ对一系列非呼吸道病毒的体外研究结果。
| Table 6 In vitro studies of TIZ against a broad spectrum of non-respiratory viruses[92] |
吩噁嗪骨架广泛用于稳定核酸双链, 用于构建荧光探针分子, 以研究核酸结构、识别和代谢等[99]。2011年, Gardarsson等[100]用吩噁嗪核苷作为探针鉴定了双链DNA的错配。此外, Iwata和Hayashi等[101, 102]发现吩噁嗪衍生物表现出对脊髓灰质炎病毒(poliovirus) (EC50值为0.25 μg·mL-1)、HSV-1 (EC50值为6.1 μg·mL-1, CC50值为41 μg·mL-1)、HSV-2 (EC50值为2.4 μg·mL-1, CC50值为41 μg·mL-1)的抗病毒活性。吩噁嗪核苷的抗病毒作用机制为掺入核酸链中并损害细胞和病毒的复制和转录, 影响细胞或病毒聚合酶合成DNA和RNA的效率[103]。
Kozlovskaya等[99]设计并合成了一系列带有2'-脱氧核糖或核糖基团的吩噁嗪核苷衍生物, 并测定了它们对一组结构多样的DNA和RNA病毒的抗病毒活性。相关吩噁嗪衍生物的结构如图 4所示, 其抗病毒活性结果列于表 7中。

|
Figure 4 The structures of some phenoxazine nucleosides |
| Table 7 EC50 (μmol·L-1) and CC50 (μmol·L-1) of phenoxazine nucleosides against herpesvirus[99] |
化合物22a、25a、26a、30a和34a显示出对VZV的活性。其中化合物22a是针对VZV OKA (TK+)毒株活性最好的化合物, 其EC50为0.06 μmol·L-1, 但它对07-1 (TK-)毒株的活性大幅降低, 其EC50约为10 μmol·L-1。与对照药物相比, 22a对OKA毒株的活性是阿昔洛韦的42倍, 是布列夫定的1/3。与对照药物相似, 化合物22a对TK缺陷型VZV的活性降低。化合物25a、26a和33a显示出在微摩尔水平内的抗VZV活性, EC50值分别为1.5 ± 0.3、1.5 ± 0.3和1.7 ± 0.1 μmol·L-1, 其对人胚胎肺细胞(human embryonic lung cells, HEL)的CC50值分别为12.2 ± 0.8、9.7 ± 1.5和14.4 ± 0.7 μmol·L-1, 并且证明对VZV TK+和TK-毒株具有相同的活性, 它们对OKA毒株的EC50值与阿昔洛韦(EC50 = 2.5 ± 0.4 μmol·L-1)相当。25a、27a、31a、32a和33a仅显示针对CMV Davis毒株的微弱活性, 其EC50值分别为4、2.9、2.19、10.9和1.27 μmol·L-1, 与更昔洛韦(EC50 = 1.06 μmol·L-1)相当, 远远低于西多福韦(EC50 = 0.3 μmol·L-1)[99]。
3.4 绕丹宁和硫代巴比妥类广谱抗病毒药物Cagno等[1]最近发现了一些绕丹宁和硫代巴比妥类衍生物(39~45)对HSV-1或HSV-2具有活性, 而它们对无包膜病毒HPV是完全无效的。同时已有研究证明这类化合物对包膜病毒的作用机制是改变半融合茎中的融合动力学, 进而影响病毒包膜的正常形成[104, 105]。用空斑效率测定法测试化合物对不同病毒的活性, 发现化合物42具有较好的抗病毒活性, 其对HSV-1、HSV-2和HCMV的EC50值分别为2.18、1.60和4.22 nmol·L-1, CC50值分别为2.09、2.09和5.24 nmol·L-1。测定了化合物42针对包膜病毒和无包膜病毒的抗病毒活性谱(表 8)。在非细胞毒性剂量下, 化合物42对测试的所有包膜病毒均有抑制活性, 其EC50值为1.60~69.9 nmol·L-1, CC50值为2.09~135.7 nmol·L-1, 对无包膜病毒均无活性, 表明其作用机制可能是影响病毒包膜的正常形成[1]。
| Table 8 EC50 (nmol·L-1), EC90 (nmol·L-1) and CC50 (nmol·L-1) of 42[1] |
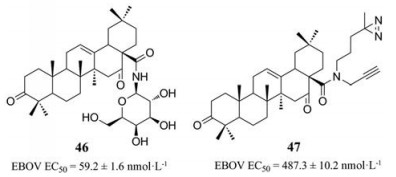
三萜类是一类植物次生代谢产物, 通常被认为是植物防御病原体感染的主要成分, 其结构特征为疏水性五元环。细胞研究已经证实三萜天然产物齐墩果酸(oleanolic acid, OA)和白桦脂酸(betulinic acid, BA)具有抗HIV作用[106]。此外, 还发现OA和另一种三萜天然产物合欢酸(echinocystic acid, EA)的类似物或衍生物对Ⅳ和HCV复制具有抑制活性, 且活性与其化学结构密切相关[107, 108]。在此基础上, 北京大学周德敏课题组发现三萜类化合物是EBOV和马尔堡病毒(Marburg virus, MARV)的侵入抑制剂, 从而扩大了三萜类化合物作为侵入抑制剂的抗病毒谱。其中代表性化合物是46与47, 抑制EBOV的EC50分别是59.2和487.3 nmol·L-1 [109]。
|
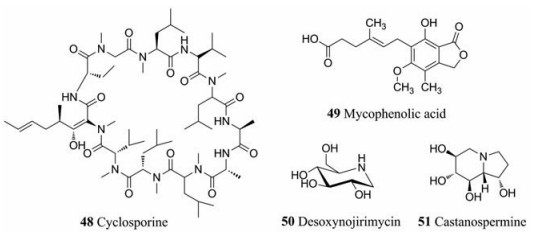
环孢菌素A (48, CsA)是从真菌中分离的具有免疫抑制活性的环肽[110]。进一步的研究发现, 细胞亲环素与HIV Gag多聚蛋白和牛痘病毒晚期蛋白之间存在着相互作用, 可以促进复制复合物的正确折叠和组装。环孢菌素A的抗病毒作用机制是与亲环素结合进而阻断这些相互作用, 从而阻断病毒复制[5]。此外, 环孢菌素A可通过类似的机制抑制HPV、HBV、HCV、VSV、HCMV、流感病毒、冠状病毒等的复制[9, 62, 111, 112]。
|
霉酚酸(49, mycophenolic acid, MPA)通过干扰嘌呤从头合成来阻断核酸合成[113]。首次观察到MPA限制了牛痘病毒(vaccinia virus, VV)和HSV在细胞培养基中的细胞病变效应[114]。后续研究表明, 它可以通过影响病毒核酸合成来抑制DENV、WNV、HBV、HCV、HIV、戊型肝炎病毒(hepatitis E virus, HEV)、汉坦河病毒(Hantaan river virus)和痘病毒(poxviruses)的复制[115-121]。
1-脱氧野尻霉素(50, 1-deoxynojirimycin, DNJ)和栗精胺(51, castanospermine, CST)的衍生物西戈斯韦(celgosivir)已被确认为在体内外抑制多种病毒的复制[122-124]。在哺乳动物细胞中, 这两个化合物均可以阻断内质网α-葡糖苷酶Ⅰ和Ⅱ的功能。α-葡糖苷酶Ⅰ和Ⅱ负责修剪在内质网蛋白质合成过程中添加到N-连接聚糖中的葡萄糖残基[125]。葡萄糖残基的添加步骤是糖蛋白成熟过程中提高蛋白质折叠效率的中间步骤。因此, CST和DNJ的作用机制被认为是干扰了一些病毒糖蛋白的折叠过程, 导致成熟包膜难以形成[126]。
4 总结与展望尽管药物化学家已经付出了艰苦卓绝的努力, 仍有艾滋病、乙肝等棘手的感染性疾病未被根治。此外, 新发病毒疫情对人类的健康与生命构成了持续性的威胁。例如, 大约100年前, 非洲猪瘟病毒(African swine fever virus, ASFV)就在非洲肯尼亚被首次确定, 而在2018年8月3日, 中国农业农村部新闻办公室通报我国首例非洲猪瘟疫情, 随后病毒很快传播到全国大部分地区, 成为了公共健康领域的重大问题。加之病毒自身的快速突变特性, 引起耐药突变毒株的流行, 使防控形势非常严峻。自SARS-CoV-2感染疫情暴发以来, 相关部门即出台相应政策支持相关治疗药物研发。众所周知抗病毒药物的研发通常需要大量的资源和时间, 尽管目前具有药物研发的软硬件条件, 但在应对此次突发性的、高传染性的病原体所造成的公共卫生危机时, 显然“远水不及近渴”。因此老药新用成为新近针对SARS-CoV-2药物研发的首选策略。特别是优先考虑已知的对SRAS-CoV和MERS-CoV广谱抗冠状病毒药物及靶向宿主的广谱抗病毒药物。但是, 目前可用的广谱抗病毒药物的种类和数量依然稀少, 因此, 迫切需要强化广谱抗病毒药物研究。
|
在本综述中, 笔者重点介绍了针对病毒生命周期中特定靶标和具有独特优势结构的广谱抗病毒药物的最新进展, 涵盖靶向聚合酶和病毒包膜、宿主蛋白的广谱抗病毒药物, 以及具有特定优势结构的广谱抗病毒药物。基于化合物的表型筛选与靶标确证是发现全新骨架的广谱抗病毒药物的重要途径。表型药物筛选是通过测试化合物对复杂生物体系及相关特异性靶分子和/或信号通路的改变, 发现改变生物表型的药物[127]。与此同时化学蛋白质组学和计算机技术辅助等靶点确认技术的发展可以弥补表型筛选在靶点确证方面的不足, 进而推动发现全新骨架全新机制的广谱抗病毒药物。表型筛选的基础是有足够多结构类型不同的化合物, 因此构建结构类型不同的化合物库是进行化合物表型筛选的首要工作。微量合成等化合物库构建新技术可以实现在短时间内获得大量结构不同的化合物, 与原位筛选等筛选方法相结合, 可以实现短时间内对大量结构不同的化合物进行合成与表型筛选, 进而加速苗头化合物的发现过程。
此外, 在广谱抗病毒药物的结构优化中涉及到一些典型的药物化学策略, 如药物再定位、分子杂合、基于片段的药物发现、基于结构的化合物优化以及骨架跃迁、生物电子等排等策略。综合使用这些策略可以在药物开发早期阶段节省时间与费用, 可以加速先导化合物的发现、优化过程, 进而加快广谱抗病毒药物的开发过程。
需要注意的是, 作用于宿主细胞的广谱抗病毒药物的主要缺点是体外抑制活性与体内治疗效果的不完全一致, 其原因可能是机体系统性机制可以代偿被阻断的靶标效应[5]。针对这一问题, 可以通过确证对于病毒复制至关重要但对宿主细胞无关紧要的新靶标来解决; 还可以设计多靶点广谱抗病毒药物, 使其可以作用于该通路上尽可能多的相关靶点, 进而达到抑制该靶点效应的目的。
总之, 广泛使用新颖的药物化学策略及化合物库合成与筛选新技术, 可以大大加速广谱抗病毒药物的开发进程。
| [1] |
Cagno V, Tintori C, Civra A, et al. Novel broad spectrum virucidal molecules against enveloped viruses[J]. PLoS One, 2018, 13: 12. |
| [2] |
Howard CR, Fletcher NF. Emerging virus diseases: can we ever expect the unexpected[J]. Emerg Microbes Infect, 2012, 1: 1-9. |
| [3] |
Ianevski A, Andersen PI, Merits A, et al. Expanding the activity spectrum of antiviral agents[J]. Drug Discov Today, 2019, 24: 1224-1228. DOI:10.1016/j.drudis.2019.04.006 |
| [4] |
Ianevski A, Zusinaite E, Kuivanen S, et al. Novel activities of safe-in-human broad-spectrum antiviral agents[J]. Antiviral Res, 2018, 154: 174-182. |
| [5] |
Martinez JP, Sasse F, Bronstrup M, et al. Antiviral drug discovery: broad-spectrum drugs from nature[J]. Nat Prod Rep, 2015, 1: 29-48. |
| [6] |
To KKW, Zhou J, Chan JFW, et al. Host genes and influenza pathogenesis in humans: an emerging paradigm[J]. Curr Opin Virol, 2015, 14: 7-15. DOI:10.1016/j.coviro.2015.04.010 |
| [7] |
Alaqeel SI. Synthetic approaches to benzimidazoles from o-phenylenediamine: a literature review[J]. J Saudi Chem Soc, 2017, 2: 229-237. |
| [8] |
Brai A, Fazi R, Tintori C, et al. Human DDX3 protein is a valuable target to develop broad spectrum antiviral agents[J]. Proc Natl Acad Sci U S A, 2016, 113: 5388-5393. |
| [9] |
Lin K, Gallay P. Curing a viral infection by targeting the host: the example of cyclophilin inhibitors[J]. Antiviral Res, 2013, 99: 68-77. DOI:10.1016/j.antiviral.2013.03.020 |
| [10] |
Zeisel MB, Lupberger J, Fofana I, et al. Host-targeting agents for prevention and treatment of chronic hepatitis C-perspectives and challenges[J]. J Hepatol, 2013, 58: 375-384. |
| [11] |
Zhu JD, Meng W, Wang XJ, et al. Broad-spectrum antiviral agents[J]. Front Microbiol, 2015, 6: 517. |
| [12] |
Garcia-Serradilla M, Risco C, Pacheco B. Drug repurposing for new, efficient, broad spectrum antivirals[J]. Virus Res, 2019, 264: 22-31. |
| [13] |
Ng KK, Arnold JJ, Cameron CE. Structure-function relationships among RNA-dependent RNA polymerases[J]. Curr Top Microbiol Immunol, 2008, 320: 137-156. |
| [14] |
Liu H, Naismith JH, Hay RT. Adenovirus DNA replication[J]. Curr Top Microbiol Immunol, 2003, 272: 131-164. |
| [15] |
Chang KO, George DW. Interferons and ribavirin effectively inhibit Norwalk virus replication in replicon-bearing cells[J]. J Virol, 2007, 81: 12111-12118. |
| [16] |
Snell NJC. Ribavirin-current status of a broad spectrum antiviral agent[J]. Expert Opin Pharmacother, 2001, 8: 1317-1324. |
| [17] | |
| [18] |
Sofia MJ, Chang W, Furman PA, et al. Nucleoside, nucleotide, and non-nucleoside inhibitors of hepatitis C virus NS5B RNA-dependent RNA-polymerase[J]. J Med Chem, 2012, 55: 2481-2531. DOI:10.1021/jm201384j |
| [19] |
Siegel D, Hui HC, Doerffler E, et al. Discovery and synthesis of a phosphoramidate prodrug of a pyrrolo [2, 1-f][triazin-4-amino] adenine C-nucleoside (GS-5734) for the treatment of Ebola and Emerging viruses[J]. J Med Chem, 2017, 60: 1648-1661. DOI:10.1021/acs.jmedchem.6b01594 |
| [20] |
Tchesnokov EP, Feng JY, Porter DP, et al. Mechanism of inhibition of Ebola virus RNA-dependent RNA polymerase by remdesivir[J]. Viruses, 2019, 11: 326. |
| [21] |
Lo MK, Jordan R, Arvey A, et al. GS-5734 and its parent nucleoside analog inhibit Filo-, Pneumo-, and Paramyxoviruses[J]. Sci Rep, 2017, 7: 43395. |
| [22] |
Sheahan TP, Sims AC, Graham RL, et al. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses[J]. Sci Transl Med, 2017, 9: 396. |
| [23] |
Warren TK, Jordan R, Lo MK, et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys[J]. Nature, 2016, 531: 381-385. DOI:10.1038/nature17180 |
| [24] |
Wang M, Cao R, Zhang L, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro[J]. Cell Res, 2020. DOI:10.1038/s41422-020-0282-0 |
| [25] |
Anthony SJ, Epstein JH, Murray KA, et al. A strategy to estimate unknown viral diversity in mammals[J]. MBio, 2013, 4: 13. |
| [26] |
Wisskirchen K, Lucifora J, Michler T, et al. New pharmacological strategies to fight enveloped viruses[J]. Trends Pharmacol Sci, 2014, 9: 470-478. |
| [27] |
Mackenzie JS, Jeggo M. Reservoirs and vectors of emerging viruses[J]. Curr Opin Virol, 2013, 2: 170-179. |
| [28] |
McMahon HT, Gallop JL. Membrane curvature and mechanisms of dynamic cell membrane remodelling[J]. Nature, 2005, 438: 590-596. DOI:10.1038/nature04396 |
| [29] |
Chen M, Aoki-Utsubo C, Kameoka M, et al. Broad-spectrum antiviral agents: secreted phospholipase A2 targets viral envelope lipid bilayers derived from the endoplasmic reticulum membrane[J]. Sci Rep, 2017, 7: 1-8. DOI:10.1038/s41598-016-0028-x |
| [30] |
Shivanna V, Kim Y, Chang KO. Endosomal acidification and cathepsin L activity is required for calicivirus replication[J]. Virology, 2014, 464: 287-295. |
| [31] |
Chandran K, Sullivan NJ, Felbor U, et al. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection[J]. Science, 2005, 308: 1643-1645. DOI:10.1126/science.1110656 |
| [32] |
Rolain JM, Colson P, Raoult D. Recycling of chloroquine and its hydroxyl analogue to face bacterial, fungal and viral infections in the 21st century[J]. Int J Antimicrob Agents, 2007, 30: 297-308. DOI:10.1016/j.ijantimicag.2007.05.015 |
| [33] |
Salata C, Calistri A, Parolin C, et al. Antiviral activity of cationic amphiphilic drugs[J]. Expert Rev Anti Infect Ther, 2017, 15: 483-492. DOI:10.1080/14787210.2017.1305888 |
| [34] |
Savarino A, Boelaert JR, Cassone A, et al. Effects of chloroquine on viral infections: an old drug against today's diseases[J]. Lancet Infect Dis, 2003, 3: 722-727. DOI:10.1016/S1473-3099(03)00806-5 |
| [35] |
Brouwers J, Vermeire K, Schols D, et al. Development and in vitro evaluation of chloroquine gels as microbicides against HIV-1 infection[J]. Virology, 2008, 378: 306-310. |
| [36] |
de Wilde AH, Jochmans D, Posthuma CC, et al. Screening of an FDA-approved compound library identifies four small-molecule inhibitors of Middle East respiratory syndrome coronavirus replication in cell culture[J]. Antimicrob Agents Chemother, 2014, 58: 4875-4884. |
| [37] |
Kaptein SJF, Neyts J. Towards antiviral therapies for treating dengue virus infections[J]. Curr Opin Pharmacol, 2016, 30: 1-7. DOI:10.1016/j.coph.2016.06.002 |
| [38] |
Madrid PB, Panchal RG, Warren TK, et al. Evaluation of Ebola virus inhibitors for drug repurposing[J]. ACS Infect Dis, 2015, 1: 317-326. DOI:10.1021/acsinfecdis.5b00030 |
| [39] |
Delvecchio R, Higa LM, Pezzuto P, et al. Chloroquine, an endocytosis blocking agent, inhibits Zika virus infection in different cell models[J]. Viruses, 2016, 8: 322. |
| [40] |
Cordin O, Banroques J, Tanner NK, et al. The DEAD-box protein family of RNA helicases[J]. Gene, 2006, 367: 17-37. DOI:10.1016/j.gene.2005.10.019 |
| [41] |
Linder P. Dead-box proteins: a family affair—active and passive players in RNP-remodeling[J]. Nucleic Acids Res, 2006, 34: 4168-4180. DOI:10.1093/nar/gkl468 |
| [42] |
Schröder M. Human DEAD-box protein 3 has multiple functions in gene regulation and cell cycle control and is a prime target for viral manipulation[J]. Biochem Pharmacol, 2010, 3: 297-306. |
| [43] |
Ricardo SR, Théophile O. The role of the DEAD‐box RNA helicase DDX3 in mRNA metabolism[J]. Wiley Interdiscip Rev RNA, 2013, 4: 369-385. |
| [44] |
Riva V, Maga G. From the magic bullet to the magic target: exploiting the diverse roles of DDX3X in viral infections and tumorigenesis[J]. Future Med Chem, 2019, 11: 1357-1381. |
| [45] |
Ariumi Y. Multiple functions of DDX3 RNA helicase in gene regulation, tumorigenesis, and viral infection[J]. Front Genet, 2014, 5: 423. |
| [46] |
Valiente‐Echeverría F, Hermoso MA, Ricardo SR. RNA helicase DDX3: at the crossroad of viral replication and antiviral immunity[J]. Rev Med Virol, 2015, 5: 286-299. |
| [47] |
Thulasi Raman SN, Liu G, Pyo HM, et al. DDX3 interacts with influenza A virus NS1 and NP proteins and exerts antiviral function through regulation of stress granule formation[J]. J Virol, 2016, 7: 3661-3675. |
| [48] |
Fazi R, Tintori C, Brai A, et al. Homology model-based virtual screening for the identification of human helicase DDX3 inhibitors[J]. J Chem Inf Model, 2015, 11: 2443-2454. |
| [49] |
Radi M, Falchi F, Garbelli A, et al. Discovery of the first small molecule inhibitor of human DDX3 specifically designed to target the RNA binding site: towards the next generation HIV-1 inhibitors[J]. Bioorg Med Chem Lett, 2012, 22: 2094-2098. DOI:10.1016/j.bmcl.2011.12.135 |
| [50] |
Harak C, Lohmann V. Ultrastructure of the replication sites of positive-strand RNA viruses[J]. Virology, 2015, 479: 418-433. |
| [51] |
Dorobantu CM, Albulescu L, Harak C, et al. Modulation of the host lipid landscape to promote RNA virus replication: the picornavirus encephalomyocarditis virus converges on the pathway used by hepatitis C virus[J]. PLoS Pathog, 2015, 11: 9. |
| [52] |
Boura E, Nencka R. Phosphatidylinositol 4-kinases: function, structure, and inhibition[J]. Exp Cell Res, 2015, 2: 136-145. |
| [53] |
Minogue S, Waugh MG. The phosphatidylinositol 4-kinases: don't call it a comeback[J]. Subcell Biochem, 2012, 58: 1-24. |
| [54] |
Mejdrova I, Chalupska D, Plackova P, et al. Rational design of novel highly potent and selective phosphatidylinositol 4-Kinase Ⅲbeta(PI4KB) inhibitors as broad-spectrum antiviral agents and tools for chemical biology[J]. J Med Chem, 2017, 60: 100-118. |
| [55] |
Knight ZA, Gonzalez B, Feldman ME, et al. A pharmacological map of the PI3-K family defines a role for p110α in insulin signaling[J]. Cell, 2006, 4: 733-747. |
| [56] |
Mejdrová I, Chalupská D, Kögler M, et al. Highly selective phosphatidylinositol 4-kinase Ⅲβ inhibitors and structural insight into their mode of action[J]. J Med Chem, 2015, 58: 3767-3793. |
| [57] |
Ahmed-Belkacem A, Colliandre L, Ahnou N, et al. Fragment-based discovery of a new family of non-peptidic small-molecule cyclophilin inhibitors with potent antiviral activities[J]. Nat Commun, 2016, 7: 1-11. |
| [58] |
Fischer G, Wittmann-Liebold B, Lang K, et al. Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins[J]. Nature, 1989, 337: 476-478. DOI:10.1038/337476a0 |
| [59] |
Takahashi N, Hayano T, Suzuki M. Peptidyl-prolyl cis-trans isomerase is the cyclosporin A-binding protein cyclophilin[J]. Nature, 1989, 337: 473-475. DOI:10.1038/337473a0 |
| [60] |
Hartono C, Muthukumar T, Suthanthiran M. Immunosuppressive drug therapy[J]. Cold Spring Harb Perspect Med, 2013, 3: 9. |
| [61] |
Bienkowska-Haba M, Patel HD, Sapp M. Target cell cyclophilins facilitate human papillomavirus type 16 infection[J]. PLoS Pathog, 2009, 5: 7. |
| [62] |
Bose S, Mathur M, Bates P, et al. Requirement for cyclophilin A for the replication of vesicular stomatitis virus New Jersey serotype[J]. J Gen Virol, 2003, 7: 1687-1699. |
| [63] |
Franke EK, Yuan HEH, Luban J. Specific incorporation of cyclophilin A into HIV-1 virions[J]. Nature, 1994, 372: 359-362. DOI:10.1038/372359a0 |
| [64] |
Hansson MJ, Moss SJ, Bobardt M, et al. Bioengineering and semisynthesis of an optimized cyclophilin inhibitor for treatment of chronic viral infection[J]. Chem Biol, 2015, 2: 285-292. |
| [65] |
Medzhitov R. Recognition of microorganisms and activation of the immune response[J]. Nature, 2007, 449: 819-826. |
| [66] |
Du K, Liu J, Broering R, et al. Recent advances in the discovery and development of TLR ligands as novel therapeutics for chronic HBV and HIV infections[J]. Expert Opin Drug Discov, 2018, 7: 661-670. |
| [67] |
Nosratabadi R, Alavian SM, Zare-Bidaki M, et al. Innate immunity related pathogen recognition receptors and chronic hepatitis B infection[J]. Mol Immunol, 2017, 90: 64-73. |
| [68] |
Gay NJ, Gangloff M. Structure and function of Toll receptors and their ligands[J]. Annu Rev Biochem, 2007, 76: 141-165. |
| [69] |
Gibbert K, Dietze KK, Zelinskyy G, et al. Polyinosinic-polycytidylic acid treatment of Friend retrovirus-infected mice improves functional properties of virus-specific T cells and prevents virus-induced disease[J]. J Immunol, 2010, 10: 6179-6189. |
| [70] |
Olbrich ARM, Schimmer S, Heeg K, et al. Effective postexposure treatment of retrovirus-induced disease with immunostimulatory DNA containing CpG motifs[J]. J Virol, 2002, 36: 11397-11404. |
| [71] |
Wang Y, Abel K, Lantz K, et al. The Toll-like receptor 7(TLR7) agonist, imiquimod, and the TLR9 agonist, CpG ODN, induce antiviral cytokines and chemokines but do not prevent vaginal transmission of simian immunodeficiency virus when applied intravaginally to rhesus macaques[J]. J Virol, 2005, 22: 14355-14370. |
| [72] |
Evans DR, Guy HI. Mammalian pyrimidine biosynthesis: fresh insights into an ancient pathway[J]. J Biol Chem, 2004, 32: 33035-33038. |
| [73] |
Chen S, Ding S, Yin Y, et al. Suppression of pyrimidine biosynthesis by targeting DHODH enzyme robustly inhibits rotavirus replication[J]. Antiviral Res, 2019, 167: 35-44. DOI:10.1016/j.antiviral.2019.04.005 |
| [74] |
Reis RAG, Calil FA, Feliciano PR, et al. The dihydroorotate dehydrogenases: past and present[J]. Arch Biochem Biophys, 2017, 632: 175-191. |
| [75] |
Munier-Lehmann HLN, Vidalain PO, Tangy FDR, et al. On dihydroorotate dehydrogenases and their inhibitors and uses[J]. J Med Chem, 2013, 56: 3148-3167. |
| [76] |
Hoffmann HH, Kunz A, Simon VA, et al. Broad-spectrum antiviral that interferes with de novo pyrimidine biosynthesis[J]. Proc Natl Acad Sci U S A, 2011, 14: 5777-5782. |
| [77] |
Luthra P, Naidoo J, Pietzsch CA, et al. Inhibiting pyrimidine biosynthesis impairs Ebola virus replication through depletion of nucleoside pools and activation of innate immune responses[J]. Antiviral Res, 2018, 158: 288-302. DOI:10.1016/j.antiviral.2018.08.012 |
| [78] |
Tan YH, Driscoll JS, Mui SM. Dihydroorotate dehydrogenase inhibitors for the treatment of viral-mediated diseases: US, 6841561 [P]. 2005-01-11.
|
| [79] |
Wang QY, Bushell S, Qing M, et al. Inhibition of dengue virus through suppression of host pyrimidine biosynthesis[J]. J Virol, 2011, 13: 6548-6556. |
| [80] |
Chung DH, Golden JE, Adcock RS, et al. Discovery of a broad-spectrum antiviral compound that inhibits pyrimidine biosynthesis and establishes a type 1 interferon-independent antiviral state[J]. Antimicrob Agents Chemother, 2016, 60: 4552-4562. DOI:10.1128/AAC.00282-16 |
| [81] |
Lucas-Hourani M, Dauzonne D, Jorda P, et al. Inhibition of pyrimidine biosynthesis pathway suppresses viral growth through innate immunity[J]. PLoS Pathog, 2013, 9: 10. |
| [82] |
Wang Y, Wang W, Xu L, et al. Cross talk between nucleotide synthesis pathways with cellular immunity in constraining hepatitis E virus replication[J]. Antimicrob Agents Chemother, 2016, 60: 2834-2848. |
| [83] |
Yang Y, Cao L, Gao H, et al. Discovery, optimization, and target identification of novel potent broad-spectrum antiviral inhibitors[J]. J Med Chem, 2019, 62: 4056-4073. |
| [84] |
Adler TK, Albert A. The biological and physical properties of the azaindoles[J]. J Med Chem, 1963, 5: 480-483. |
| [85] |
Enumula S, Pangal A, Gazge M, et al. Diverse pharmacological aspects of benzimidazole derivatives: a review[J]. Res J Chem Sci, 2014, 4: 78-88. |
| [86] |
Tonelli M, Novelli F, Tasso B, et al. Antiviral activity of benzimidazole derivatives. Novel anti-CVB-5, anti-RSV and anti-Sb-1 agents[J]. Bioorg Med Chem, 2014, 17: 4893-4909. |
| [87] |
Burgeson JR, Moore AL, Gharaibeh DN, et al. Discovery and optimization of potent broad-spectrum arenavirus inhibitors derived from benzimidazole and related heterocycles[J]. Bioorg Med Chem Lett, 2013, 23: 750-756. |
| [88] |
Cohen SA. Use of nitazoxanide as a new therapeutic option for persistent diarrhea: a pediatric perspective[J]. Curr Med Res Opin, 2005, 7: 999-1004. |
| [89] |
Fox LM, Saravolatz LD. Nitazoxanide: a new thiazolide antiparasitic agent[J]. Clin Infect Dis, 2005, 8: 1173-1180. |
| [90] |
Rossignol JF, Frazia SL, Chiappa L, et al. Thiazolides, a new class of anti-influenza molecules targeting viral hemagglutinin at the post-translational level[J]. J Biol Chem, 2009, 43: 29798-29808. |
| [91] |
Broekhuysen J, Stockis A, Lins RL, et al. Nitazoxanide: pharmacokinetics and metabolism in man[J]. Int J Clin Pharmacol Ther, 2000, 8: 387-394. |
| [92] |
Rossignol JF. Nitazoxanide: a first-in-class broad-spectrum antiviral agent[J]. Antiviral Res, 2014, 110: 94-103. DOI:10.1016/j.antiviral.2014.07.014 |
| [93] |
De Carvalho LP, Darby CM, Rhee KY, et al. Nitazoxanide disrupts membrane potential and intra-bacterial pH homeostasis of Mycobacterium tuberculosis[J]. ACS Med Chem Lett, 2011, 2: 849-854. DOI:10.1021/ml200157f |
| [94] |
Jasenosky LD, Cadena C, Mire CE, et al. The FDA-approved oral drug nitazoxanide amplifies host antiviral responses and inhibits Ebola virus[J]. iScience, 2019, 19: 1279-1290. |
| [95] |
Rossignol JF, El-Gohary YM. Nitazoxanide in the treatment of viral gastroenteritis: a randomized double-blind placebo-controlled clinical trial[J]. Aliment Pharmacol Ther, 2006, 10: 1423-1430. |
| [96] |
Frazia SL, Ciucci A, Arnoldi F, et al. Thiazolides, a new class of antiviral agents effective against rotavirus infection, target viral morphogenesis, inhibiting viroplasm formation[J]. J Virol, 2013, 20: 11096-11106. |
| [97] |
Korba BE, Montero AB, Farrar K, et al. Nitazoxanide, tizoxanide and other thiazolides are potent inhibitors of hepatitis B virus and hepatitis C virus replication[J]. Antiviral Res, 2008, 77: 56-63. |
| [98] |
Shi Z, Wei J, Deng X, et al. Nitazoxanide inhibits the replication of Japanese encephalitis virus in cultured cells and in a mouse model[J]. Virol J, 2014, 11: 10. |
| [99] |
Kozlovskaya LI, Andrei G, Orlov AA, et al. Antiviral activity spectrum of phenoxazine nucleoside derivatives[J]. Antiviral Res, 2019, 163: 117-124. |
| [100] |
Gardarsson H, Kale AS, Sigurdsson ST. Structure-function relationships of phenoxazine nucleosides for identification of mismatches in duplex DNA by fluorescence spectroscopy[J]. Chembiochem, 2011, 4: 567-575. |
| [101] |
Iwata A, Yamaguchi T, Sato K, et al. Suppression of proliferation of poliovirus and porcine parvovirusbynovel phenoxazines, 2-amino-4, 4 alpha-dihydro-4 alpha-7-dimethyl-3H-phenoxazine-3-one and 3-amino-1, 4 alpha-dihydro-4 alpha-8-dimethyl-2H-phenoxazine-2-one[J]. Biol Pharm Bull, 2005, 5: 905-907. |
| [102] |
Hayashi K, Hayashi T, Tomoda A. Phenoxazine derivatives inactivate human cytomegalovirus, herpes simplex virus-1, and herpes simplex virus-2 in vitro[J]. J Pharmacol Sci, 2008, 3: 369-375. |
| [103] |
Edwards TE, Cekan P, Reginsson GW, et al. Crystal structure of a DNA containing the planar, phenoxazine-derived bi-functional spectroscopic probe C[J]. Nucleic Acids Res, 2011, 10: 4419-4426. |
| [104] |
Tintori C, Iovenitti G, Ceresola ER, et al. Rhodanine derivatives as potent anti-HIV and anti-HSV microbicides[J]. PLoS One, 2018, 13: 6. |
| [105] |
Speerstra S, Chistov AA, Proskurin GV, et al. Antivirals acting on viral envelopes via biophysical mechanisms of action[J]. Antiviral Res, 2018, 149: 164-173. |
| [106] |
Mayaux JF, Bousseau A, Pauwels R, et al. Triterpene derivatives that block entry of human immunodeficiency virus type 1 into cells[J]. Proc Natl Acad Sci U S A, 1994, 9: 3564-3568. |
| [107] |
Yu M, Si L, Wang Y, et al. Discovery of pentacyclic triterpenoids as potential entry inhibitors of influenza viruses[J]. J Med Chem, 2014, 57: 10058-10071. DOI:10.1021/jm5014067 |
| [108] |
Yu F, Wang Q, Zhang Z, et al. Development of oleanane-type triterpenes as a new class of HCV entry inhibitors[J]. J Med Chem, 2013, 56: 4300-4319. DOI:10.1021/jm301910a |
| [109] |
Si L, Meng K, Tian Z, et al. Triterpenoids manipulate a broad range of virus-host fusion via wrapping the HR2 domain prevalent in viral envelopes[J]. Sci Adv, 2018, 4: 11. |
| [110] |
Matsuda S, Koyasu S. Mechanisms of action of cyclosporine[J]. Immunopharmacology, 2000, 47: 119-125. DOI:10.1016/S0162-3109(00)00192-2 |
| [111] |
Bienkowska-Haba M, Williams C, Kim SM, et al. Cyclophilins facilitate dissociation of the human papillomavirus type 16 capsid protein L1 from the L2/DNA complex following virus entry[J]. J Virol, 2012, 18: 9875-9887. |
| [112] |
De Wilde AH, Zevenhoven-Dobbe JC, Van Der Meer Y, et al. Cyclosporin A inhibits the replication of diverse coronaviruses[J]. J Gen Virol, 2011, 11: 2542. |
| [113] |
Kitchin JES, Pomeranz MK, Pak G, et al. Rediscovering mycophenolic acid: a review of its mechanism, side effects, and potential uses[J]. J Am Acad Dermatol, 1997, 3: 445-449. |
| [114] |
Cline JC, Nelson JD, Gerzon K, et al. In vitro antiviral activity of mycophenolic acid and its reversal by guanine-type compounds[J]. Appl Environ Microbiol, 1969, 1: 14-20. |
| [115] |
Wang Y, Zhou X, Yannick D, et al. Calcineurin inhibitors stimulate and mycophenolic acid inhibits replication of hepatitis E virus[J]. Gastroenterology, 2014, 7: 1775-1783. |
| [116] |
Chapuis AG, Rizzardi GP, Agostino C, et al. Effects of mycophenolic acid on human immunodeficiency virus infection in vitro and in vivo[J]. Nat Med, 2000, 6: 762-768. DOI:10.1038/77489 |
| [117] |
Chung DH, Sun Y, Parker WB, et al. Ribavirin reveals a lethal threshold of allowable mutation frequency for Hantaan virus[J]. J Virol, 2007, 81: 11722-11729. |
| [118] |
Diamond MS, Zachariah M, Harris E. Mycophenolic acid inhibits dengue virus infection by preventing replication of viral RNA[J]. Virology, 2002, 2: 211-221. |
| [119] |
Gong ZJ, De Meyer S, Clarysse C, et al. Mycophenolic acid, an immunosuppressive agent, inhibits HBV replication in vitro[J]. J Viral Hepat, 1999, 3: 229-236. |
| [120] |
Henry SD, Metselaar HJ, Lonsdale RCB, et al. Mycophenolic acid inhibits hepatitis C virus replication and acts in synergy with cyclosporin A and interferon-α[J]. Gastroenterology, 2006, 5: 1452-1462. |
| [121] |
Morrey JD, Smee DF, Sidwell RW, et al. Identification of active antiviral compounds against a New York isolate of West Nile virus[J]. Antiviral Res, 2002, 55: 107-116. DOI:10.1016/S0166-3542(02)00013-X |
| [122] |
Chang J, Warren TK, Zhao X, et al. Small molecule inhibitors of ER α-glucosidases are active against multiple hemorrhagic fever viruses[J]. Antiviral Res, 2013, 98: 432-440. |
| [123] |
Durantel D. Celgosivir, an α-glucosidase I inhibitor for the potential treatment of hepatitis C virus infection[J]. Curr Opin Investig Drugs, 2009, 10: 860-870. |
| [124] |
Rathore APS, Paradkar PN, Watanabe S, et al. Celgosivir treatment misfolds dengue virus NS1 protein, induces cellular pro-survival genes and protects against lethal challenge mouse model[J]. Antiviral Res, 2011, 92: 453-460. |
| [125] |
Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum[J]. Annu Rev Biochem, 2004, 1: 1019-1049. |
| [126] |
Wardrop DJ, Waidyarachchi SL. Synthesis and biological activity of naturally occurring α-glucosidase inhibitors[J]. Nat Prod Rep, 2010, 10: 1431-1468. |
| [127] |
Reisen F, Zhang X, Gabriel D, et al. Benchmarking of multivariate similarity measures for high-content screening fingerprints in phenotypic drug discovery[J]. J Biomol Screen, 2013, 10: 1284-1297. |