 2020, Vol. 55
2020, Vol. 55



登革病毒血清型1至4 (DENV-1~4)属于黄病毒科, 是一组包膜RNA病毒。黄病毒科是由节肢动物传播的疾病病原体组成, 如黄热病病毒(YFV)、日本脑炎病毒(JEV)、西尼罗河病毒(WNV)和DENV。许多黄病毒科病毒是常见的人类致病原, 具有发病率和死亡率高的特点[1]。仅DENV一项来说, 就对生活在登革热流行地区的约25亿人构成了公共卫生威胁。DENV感染常导致登革热、危及生命的登革出血热(DHF)或登革休克综合征(DSS)。全球100多个国家报道了大约50万例登革出血热和登革出血热病例, 其中每年大约有25 000人死亡, 并且登革热在我国南方沿海各省市仍有流行的趋势。但是, 目前市场上仍无特异性治疗登革病毒感染的药物[2]。因此, 研究开发低毒、高效的抗登革病毒药物具有重大意义。本文综述了抗DENV药物的研究新进展。
1 登革热病毒的结构、致病机制与临床表现 1.1 登革病毒基因组结构和功能DENV是黄病毒科的一部分, 病毒颗粒呈哑铃状、棒状或球形, 病毒颗粒外有脂蛋白包膜, 并具有包膜刺突, 病毒包膜的外层含有包膜蛋白E, 内层含有膜蛋白M, 病毒的核心部分是由病毒的单股正链RNA和病毒衣壳蛋白C共同组成的20面体核衣壳结构。登革病毒基因组是一个长度约为11 kb的单链阳性RNA, 基因组的5'端和3'端都有1个非编码区(UTR), 位于1个开放阅读框的两侧。开放阅读框编码1个长多聚蛋白, 经病毒和细胞蛋白酶的协同和翻译后加工成3个结构蛋白(衣壳[C]、前膜[prM]和包膜[E])和7个非结构蛋白(NS1、NS2A、NS2B、NS3、NS4A、NS4B和NS5) (图 1)[2]。其中, 结构蛋白参与病毒的进入和组装; 非结构蛋白参与病毒RNA复制、避先天免疫反应和病毒组装; 糖蛋白NS1在病毒RNA复制的早期阶段参与病毒RNA的复制; NS3与辅因子NS2B、RNA三磷酸酶和RNA解旋酶都发挥病毒丝氨酸蛋白酶的作用; NS5作为甲基转移酶和依赖RNA的RNA聚合酶(RdRp)发挥作用; 对于小疏水蛋白NS2A、NS4A和NS4B的功能尚不明确。这些跨膜蛋白能够将病毒复制复合体锚定在内质网(ER)膜上[3]。

|
Figure 1 Schematic diagram of DENV genome[3]. (Figure derived from Green J, et al. Annu Rep Med Chem, 2012, 47: 297-317) |
DENV感染的复制周期可分为以下几个步骤:病毒E蛋白与细胞表面受体结合、病毒通过内吞作用进入宿主细胞、病毒包膜与内体膜的融合、病毒RNA释放到细胞质中、病毒RNA基因组被翻译成多蛋白、RNA的复制、病毒在内质网上组装形成不成熟的病毒粒子以及病毒粒子从高尔基体离开细胞。DENV感染宿主细胞首先是病毒E蛋白与细胞表面受体的结合, 目前已发现多种细胞表面分子可以作为DENV的感染受体, 如氨基多糖、DC-SIGN、甘露糖受体、蛋白聚糖等[4]。然后DENV在宿主细胞受体和病毒粒子的包膜(E)蛋白的相互作用下, 通过受体介导的内吞作用进入细胞, 之后病毒在核内体中仍保持完整, 直到细胞环境变为酸性, pH的降低使病毒包膜与释放核衣壳的内体膜发生融合, 随后核衣壳被脱衣从而把病毒基因组释放到细胞质中, 这时病毒基因组发挥信使RNA的作用, 并被病毒和细胞蛋白酶翻译成多蛋白, 然后非结构蛋白复制病毒RNA, 并在内质网上发生病毒的组装, 在内质网中衣壳蛋白包裹着新合成的病毒RNA, 形成不成熟的病毒颗粒, 最后这些颗粒通过胞吐作用到达高尔基体, 并从高尔基体离开细胞(图 2)[5]。

|
Figure 2 Life cycle of dengue virus[5]. (Figure derived from Dighe SN, et al. Eur J Med Chem, 2019, 176: 431-455) |
DENV的初次感染早期症状并不明显, 容易发生误诊。典型症状为发热, 常伴乏力、肌痛、畏寒, 可能会出现皮疹、消化道症状(恶心、呕吐、腹泻)或者女性的阴道不规则出血、鼻衄以及呕血等症状。当再次感染异型DENV时, 会产生抗体依赖增强感染效应(ADE), 病症恶化为DHF, 且伴随凝血障碍、血小板减少、血管脆性和通透性增加等症状。若病情进一步加重则可能会出现DSS, 导致弥散性血管内凝血甚至威胁生命。初次感染DENV-1或DENV-3时, 会比初次感染DENV-2或DENV-4或更容易发生登革出血热和登革休克综合征, 但若再次感染DENV-2则会导致更加严重的症状, 产生DHF的风险会显著升高[6]。
2 抗DENV药物的研究进展 2.1 病毒多聚酶抑制剂登革病毒属于黄病毒科黄病毒属, HCV属黄病毒科肝炎病毒属, 两者在结构上有很大相似性, 因此基于HCV的药物研发经验, 可以将病毒多聚酶和蛋白酶作为抗登革病毒药物研究的重要靶点。
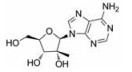
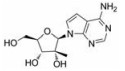
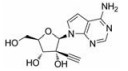
目前抑制病毒多聚酶的抑制剂有2种, 一种是核苷/核苷酸类似物, 核苷抑制剂也是应用最广泛的抗病毒药物。这些抑制剂在病毒DNA或RNA合成过程中起着链终止剂的作用[7]。例如, 黄病毒抑制剂7-去氮-甲基腺苷(MK-0608, 1), 最初研发是针对HCV的RdRp抑制剂, 而后发现其也具有抗登革病毒活性[8]; 另一个腺苷类似物7-去氮-乙炔腺苷(NITD008, 2)也能够同时在细胞水平和小鼠体内抑制登革病毒, 其三磷酸形式直接抑制登革病毒的RdRp活性, 这表明该复合物在病毒RNA合成中起着链终止子的作用; NITD008不仅能够抑制登革病毒的4种血清型, 还能够抑制黄病毒科其他病毒如WNV、黄热病毒和HCV等[9]。遗憾的是, 该化合物表现出严重的体内毒性作用。尽管如此, 对该先导化合物的结构修饰以及开发仍然是抗登革病毒药物研究领域的热点。
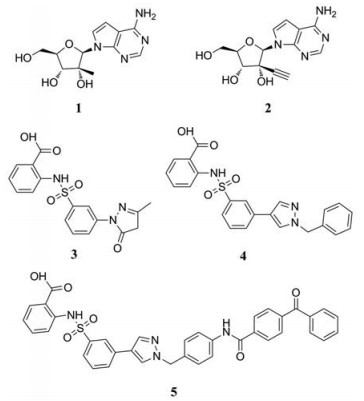
另一种抑制剂为非核苷抑制剂(non-nucleoside inhibitors, NNI), 它通过连接蛋白的变构区(allosteric pockets)来抑制酶的活性, NNI的作用机制主要是将聚合酶的结构改变成非活性构象, 阻断从聚合酶起始到延伸的构象转换或是阻碍聚合酶延伸的过程。目前已经鉴定出了一系列非核苷抑制剂[10, 11]。例如, Niyomrattanakit等[12]通过基于RdRp引物延伸的高通量筛选方法(high-throughput screening, HTS), 筛选出了3种登革病毒RdRp抑制剂, 分别是N-邻磺酰氨基苯甲酸(N-sulfonylanthranilic acid)衍生物NITD-1 (3)、NITD-2 (4)和NITD-29 (5)。它们通过与病毒NS5的RdRp域结合从而特异性的抑制登革病毒RdRp活性。
2.1.1 核苷抑制剂核苷类似物代表了一类应用最广泛的抗病毒药物, 并且一直被积极地用于DENV感染的潜在治疗。许多DENV的核苷抑制剂来源于治疗HCV药物的发现, 这是由于这两种病毒具有相似性[13]。
化合物(6) (2'-C-甲基腺苷)就是一个特别的例子, 它能抑制丙肝病毒HCV复制, 而且在2003年Migliaccio等[14]发现它也具有抗DENV活性。2'-C-甲基腺苷(6)具有抗DENV活性, 其EC50为4 µmol·L-1。但是这种化合物不适合药物开发, 因为它通过宿主脱氨酶快速转化成了2'-C-甲基肌苷。2004年Olsen等[15]用碳取代2'-C-甲基腺苷的7位氮, 减少了宿主脱氨酶对化合物的脱氨基作用, 从而发现了化合物(MK-0608, 1)。然而, 这种替代也降低了抗登革病毒活性(EC50 =15 µmol·L-1), 为了提高抗病毒活性, 2009年Yin等[9]用2'-乙炔基取代2'-C-甲基得到化合物(NITD008, 2)。2010年Chen等[16]对腺苷碱基C7位进一步修饰, 发现了化合物(NITD449, 7), 但NITD449的药代动力学性质较差。为了克服这个问题他们又合成了化合物(NITD203, 8), 以改善NITD449的药代动力学性质(表 1)。
|
| Table 1 The adenosine-based DENV nucleoside inhibitors[17]. (Data from Chen YL, et al. Antiviral Res, 2015, 122: 12-19) |
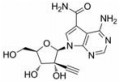
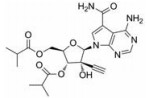
NS5蛋白是DENV基因组编码的最大最保守的蛋白, 在基因组复制、转录和加盖中起着重要作用, 且其在4种DENV血清型中高度保守(约占序列同源性的70%)。NS5有两个功能域:一个是甲基转移酶(MTase)域存在于N端; 另一个是RdRp域存在于C端。这两个功能域通过一段短的低保守性的氨基酸序列链接起来。RdRp是病毒复制周期中的关键酶。RdRp先以正链RNA为模板合成负链RNA, 然后再以后者为模板合成更多的正链RNA, 用于蛋白的翻译或病毒粒子的包装。因此它在登革病毒的生命周期中是必不可少的。
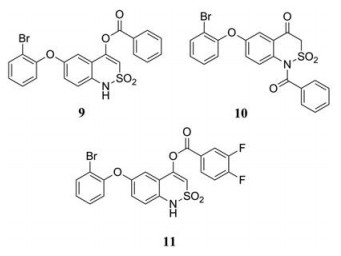
为了寻找全新的NS5 RdRp抑制剂, Dighe等[5]对其内部化合物库进行了高通量筛选, 发现了苯并噻唑嗪衍生物(9)和(10) (EC50分别为30.8和11.4 µmol·L-1)。此外, 在结构优化过程中又得到了新的苯并噻唑嗪衍生物(11) (EC50 = 0.6 ± 0.1 µmol·L-1), 不过化合物(11)在浓度为20 µmol·L-1时无抗病毒活性, 推测可能是因为(11)被代谢成相应的去苯甲酰衍生物而丧失了活性。
|
蛋白酶是产生成熟病毒所必需的, 它在维持病毒的活性方面也起着关键作用。这种酶介导多蛋白的裂解, 释放病毒繁殖所需要的功能蛋白。蛋白酶可分为NS3蛋白酶、NS2B-NS3蛋白酶及NS4B蛋白酶等, 其中双组分蛋白酶NS2B-NS3不仅负责处理登革热病毒中NS2A/NS2B、NS2B/NS3、NS3/NS4A和NS4B/NS5的连接部位, 还负责处理C、2A、NS3和NS4A的内部位点。因此, NS2B-NS3蛋白酶是药物发现的理想靶标[18]。
目前已有10个HIV蛋白酶抑制剂和2个HCV蛋白酶抑制剂用于临床, 这为研制出登革热病毒蛋白酶抑制剂提供了可能[19, 20]。然而蛋白酶抑制剂仍存在一些缺点, 例如它容易诱导耐药性病毒株的产生, 不同血清型的登革热病毒NS3蛋白酶的序列相似性在63%~74%之间, 蛋白酶抑制剂对不同亚型病毒的抑制活性也可能不同, 这都会导致耐药性病毒株的产生。因此, 登革热病毒蛋白酶抑制剂可能需要和其他药物联用。此外, 人体细胞中存在诸多丝氨酸蛋白酶、弗林蛋白酶、胰蛋白酶、凝血酶或者弹性蛋白酶等, 这可能使病毒蛋白酶抑制剂对宿主细胞蛋白酶产生交叉抑制, 这就需要抑制剂特异性抑制登革病毒丝氨酸蛋白酶活性, 同时不对宿主细胞蛋白酶产生影响[21]。
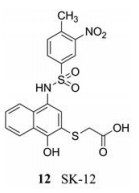
人们通过大规模化合物筛选, 发现了多种针对蛋白酶的化合物, 但是这些化合物的活性都不高, 均为微摩尔水平。比如Pambudi等[22]筛选出的小分子化合物SK-12 (12), 该化合物可在NS2B结合位点抢先与NS3结合, 干扰NS2B-NS3复合物的形成, 因此, SK-12对所有血清型的登革病毒都有抑制作用。其中, SK-12对DENV-4的抑制作用最强(EC50 = 0.74 ± 0.48 μmol·L-1)。
|
NS3蛋白酶是一种类似胰蛋白酶的丝氨酸蛋白酶, 含有典型的丝氨酸蛋白酶催化三联体, 由残基组氨酸51 (His51)、天冬氨酸75 (Asp75)和丝氨酸135 (Ser135)组成。登革病毒NS3蛋白酶(NS3pro)的N-端三分之一是蛋白酶活性所必需的, C-端三分之二则与核苷三磷酸酶和RNA解旋酶的酶活性有关。激活NS2B辅因子是NS3蛋白酶催化活性的先决条件, 双组分NS2B-NS3pro蛋白酶在功能研究和药物发现研究中比单纯的NS3pro更具有结构相关性。然而, NS2B促进NS3pro高活化的机制仍不清楚[23]。
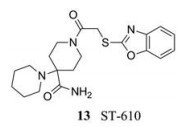
2013年, Byrd等[24]通过高通量筛选确定了一种新的NS3蛋白酶抑制剂(ST-610, 13), 在细胞培养中, 该化合物对4种血清型登革病毒均有抑制活性(EC50 = 0.203~0.272 µmol·L-1)。ST-610可能是通过抑制病毒NS3解旋酶的双链RNA (dsRNA)解离活性, 选择性地抑制DENV复制的。然而, 位于DENV NS3蛋白解旋酶区域的263位丙氨酸会突变成苏氨酸, 从而降低ST-610的敏感性使其不能抑制这一过程。因此, 目前仍需不断努力以提高这一系列化合物的抑制活性。
|
NS2B/NS3pro在登革病毒多蛋白的加工和复制过程中发挥着重要作用, 它是DENV复制过程中必不可少的酶。因此以其为靶点的药物可以抑制DENV多蛋白的加工。此外, 该蛋白酶在4种登革热病毒血清型之间高度保守, 所以NS2B-NS3蛋白酶抑制剂很可能对它们同样有效[25]。
登革病毒基因组包含1个单一的开放阅读框架(ORF), 该框架被翻译成1个独特的多蛋白, 宿主和病毒蛋白酶切割这种多蛋白, 产生3个结构蛋白(衣壳[C]、前膜[prM]和包膜[E])和7个非结构蛋白(NS1、NS2A、NS2B、NS3、NS4A、NS4B和NS5) (图 3A)。在非结构蛋白中, NS3是一种多功能蛋白复合物(图 3B), 其C端区域具有解旋酶(NS3Hel)、核苷酸-三磷酸酶(NTPase)和RNA-三磷酸酶(RTPase), 这些酶在病毒基因组复制和转录过程中都是必不可少的。而NS3的N-端区域非常不稳定, 相应的酶活性也依赖于NS2B辅助蛋白(图 3C)[26]。

|
Figure 3 Schematic diagram of DENV genome and crystal structure of NS2B/NS3pro[26]. (Derived from Leonel CA, et al. Arch Virol, 2018, 163: 575-586) |
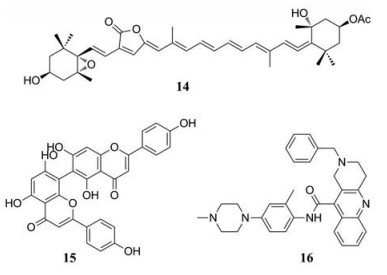
在过去的几年中, NS3pro/NS2B蛋白酶抑制剂的开发工作一直在进行, 为了开发NS3pro/NS2B蛋白酶抑制剂, 研究人员进行了小分子库的高通量筛选(HTS), 以及设计了能够模拟天然底物的肽类化合物。例如Lee等[27]从台湾珊瑚的乙醇提取物中分离出9种化合物, 并在体外测试它们抑制DENV复制的效果。在筛选的化合物中, 化合物(14)活性最强(EC50 = 4.50 ± 0.46 µmol·L-1)且化合物(14)对四种血清型的DENV均有活性。De Sousa等[28]测试了植物中的6种黄酮类化合物(长春花黄酮、槲皮苷、异槲皮苷、杨梅素、水合槲皮素和山柰酚)对NS3pro/NS2B蛋白酶的抑制活性, 其中长春花黄酮(15)是活性最好的黄酮类化合物, 它对DENV-2 (EC50 = 15.1 ± 2.2 µmol·L-1)和DENV-3 (EC50 = 25.7 ± 0.9 µmol·L-1)有比较好的抑制作用。Li等[29]通过高通量虚拟筛选(HTVS)在包含500万个分子的化学库中筛选出了14个化合物进行研究, 其中化合物(16)在300 µmol·L-1的浓度下对NS3pro/NS2B蛋白酶的抑制率为85.3%, 它是对所有血清型的DENV都有效的NS3pro/NS2B蛋白酶抑制剂(EC50 = 5.0 µmol·L-1)。
2.2.3 NS4B蛋白酶抑制剂NS4B是一种小型的膜蛋白(约27 kDa), 具有较高的疏水性。它较为保守, 在4种血清型登革热病毒(DENV-1~4)和其他黄病毒(包括YFV、WNV、JEV和TBEV)之间约有40%的氨基酸相似性。由于NS4B的疏水性, 虽然目前对其晶体结构和核磁共振结构未见报道, 但Miller等[30]利用生化分析建立了DENV-2 NS4B薄膜拓扑模型(图 4)。在这个模型中, NS4B整合在内质网(ER)膜中, 有3个跨膜结构域(TMD3、4、5)和两个膜相关结构域(pTMD1和pTMD2)。其中pTMD1和pTMD2位于NS4B的N端100个氨基酸中, 位于ER腔内。TMD3 (残基101-129)从管腔跨越到细胞质一侧的膜, 而TMD4 (残基165-190)从细胞质跨越到管腔一侧的膜。TMD5 (残基217-244)从管腔跨越到细胞质一侧的膜, 在C端被NS2B/NS3蛋白酶裂解后, 可能会翻转回内质网腔。
|

|
Figure 4 Membrane structure of NS4B and location of drug resistance mutation in NS4B[31]. (Derived from Xie X, et al. Antiviral Res, 2015, 118: 39-45) |
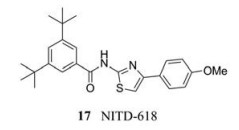
针对NS4B设计的酰胺噻唑化合物NITD-618 (17), 它是一种高效的血清型DENV抑制剂, EC50为1.0~4.1 µmol·L-1。为了测试该化合物对DENV的特异性, 研究人员对一组RNA病毒进行了NITD-618的筛选, 发现该化合物对其他经测试的RNA病毒都没有活性。值得注意的是, 酰胺噻唑耐药突变P104L和酰胺噻唑耐药突变A119T在DENV的4种血清型中都是保守的, 但在其他黄病毒中不保守, 这可能解释了NITD-618对DENV的选择性抗病毒活性。然而, NITD-618的高亲脂性导致其药代动力学性质较差, 阻碍了在对其体内药效的测试。研究人员试图降低其亲脂性, 结果却导致其活性的丧失或对DENV的抗病毒选择性的降低[32]。
2.3 病毒侵入抑制剂近年来, 人们在阐明登革病毒感染的宿主细胞途径方面取得了进展。有人提出, 登革病毒表面的病毒表位可以引发细胞免疫反应, 进而引发严重疾病。因此, 这些表位可能成为开发新型DENV侵入抑制剂的潜在靶点。
|
登革病毒侵入宿主细胞的过程如下:首先, 登革病毒包膜E蛋白与细胞膜表面的受体分子结合使得病毒颗粒吸附, 然后吸附的病毒通过受体介导的内吞进入细胞, 之后在核内体的酸性环境下病毒包膜与宿主核内体膜融合, 最后核酸衣壳进入胞质, 病毒RNA释放到胞浆(图 5)[33, 34]。宿主细胞中许多因子(包括宿主细胞表面的HS或高度硫酸化形式的糖胺聚糖)都与登革病毒的这一侵入过程有关, 如硫酸软骨素E (CSE)对于黄病毒(包括DENV)的侵入过程就是必不可少的[35]。因此, 在登革病毒侵入过程中有关的宿主因子和病毒E蛋白结构域都可能是发现抗登革病毒抑制剂的有效靶点。

|
Figure 5 Schematic diagram of DENV membrane fusion process[35]. (Derived from Hidari K, et al. Viruses, 2013, 5: 605-618) |
一些研究小组已经证明从E糖蛋白中提取的短肽可以抑制DENV的复制, 它使蛋白质结合到E蛋白三聚体中间, 诱导DENV病毒粒子表面结构变化, 从而干扰病毒和细胞结合。例如肽1 (FWFTLIKTQAKQPARYRRFC)和肽2 (MAILGDTAWDFGSLGGVFSIGKALHQVFGAIY)。肽1是一种用于置换E糖蛋白Ⅱ区铰链区域和E糖蛋白的Ⅰ区/Ⅱ区连接区域的肽, 它在病灶形成单位(FFU)检测中IC50值是7 µmol·L-1 [36]; 肽2是一种有33个氨基酸的肽, 它模仿DENV-2的茎区(残基412-144), 在恒河猴肾细胞(LLC-MK2)中进行的FFU检测证明, 它在浓度2~5 µmol·L-1内可以减少4种血清型登革热病毒50%的传染性[37]。
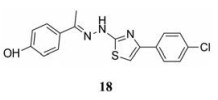
2.3.2 以E糖蛋白的疏水囊为靶点的侵入抑制剂2003年, Modis等[38]结合DENV E糖蛋白的晶体结构, 描述了结构域Ⅰ和结构域Ⅱ之间的疏水口袋, 并认为该口袋是一个有价值的配体结合位点。进而发现了靶向该位点的首个配体分子正辛酰-β-D-葡糖苷(β-OG), 因此该结合位点也被称为β-OG口袋。最近, Jadav等[39]报道了一类新的β-OG口袋混合抑制剂, 这类化合物是先前在虚拟筛选中发展而来的。其中所发现的抑制剂中化合物(18)的活性最高(EC50 = 1.39 ± 0.06 µmol·L-1, CC50 = 125 ± 40.8 µmol·L-1)。
|
E糖蛋白结构域Ⅲ似乎通过与糖胺聚糖(GAG)受体结合, 从而负责宿主细胞表面DEVN的接触和积累, GAGs是一种与靶细胞表面蛋白连接的长链而无支链的硫酸多糖。此外, DENV被证明可以与宿主细胞外表面的肝素-硫酸盐蛋白聚糖或合癸聚糖结合, 因此, 许多研究者希望通过模拟这些受体的肝素-硫酸盐(HS)部分来发现新的侵入抑制剂。
目前, 研究发现了一种可能与E糖蛋白结合的化合物, 替考拉宁衍生物(19), 该化合物体外抑制DENV-2复制的EC50为69 µmol·L-1, SI大于15。值得注意的是, 该化合物还对带有抗体的DENV颗粒发挥抗病毒作用, 因此在预防由抗体依赖增强感染效应引起的严重登革热方面具有重要价值[40]。
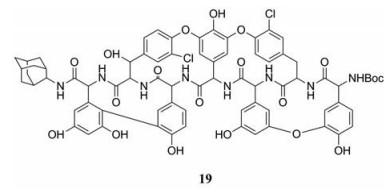
|
登革病毒是有包膜的单股正链RNA病毒, 基因组长约11 kb, 依次编码了3个结构蛋白(C、PrM和E)和7个非结构蛋白。其中衣壳蛋白C位于病毒颗粒内部, 与基因组RNA结合, 共同构成病毒的核衣壳。衣壳蛋白相对分子质量约为12 000, 是登革病毒翻译过程中首先合成的多肽, 它存在于宿主细胞的细胞核与细胞质中。登革病毒衣壳蛋白是一种具有多种生物学功能的病毒结构蛋白, 除了与病毒RNA共同构成核衣壳, 还能够与宿主细胞内诸如RNA、蛋白质等多种生物大分子相互作用, 此外它还参与了宿主细胞生长、增殖以及凋亡的调控。随着研究的不断深入, 登革病毒衣壳蛋白也成为药物研发的热点靶标[41]。
Byrd等[42]发现小分子抑制剂ST-148 (20)能够抑制DENV复制。腹膜腔内和静脉注射ST-148到AG129小鼠模型后, 能够显著降低血浆和肝脾脏中的病毒载量。SR-148耐药实验结果发现, C蛋白S34L位点的单氨基酸突变后, DENV-2对ST-148的敏感性降低了550倍, 且体外实验证明ST-148能够与野生型和突变型的C蛋白结合, 由此得出结论C蛋白是ST-148的作用靶点。Scaturro等[43]进一步研究证实ST-148提高了C蛋白分子间的作用, 导致核衣壳结构刚性化, 干扰了病毒核衣壳的组装及释放。
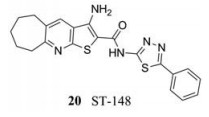
|
登革病毒在感染和发病期间宿主和病毒会发生相互作用, 登革病毒感染的宿主细胞包括单核细胞、巨噬细胞和树突状细胞。病毒通过E蛋白介导附着在细胞表面, 并通过受体介导的内吞作用进入细胞。内涵体中较低pH值触发了E蛋白的结构重组, 从而诱导了病毒和宿主细胞膜的融合, 使核衣壳和病毒RNA释放到细胞质中[44, 45]。病毒复制周期中需要多个宿主因子参与, 干扰这些宿主因子的功能往往可以抑制病毒的复制。病毒的复制依赖于宿主提供核苷。因此参与核苷生物合成的宿主酶有望作为抗登革病毒药物开发的潜在靶点。例如利巴韦林(21) (抗登革病毒药物), 它就是通过抑制鸟嘌呤的生物合成而抑制登革病毒的复制, 类似地, 嘧啶生物合成抑制剂可以通过抑制嘧啶的生物合成来抑制登革病毒的复制, 也可以作为潜在抗登革病毒药物开发的靶点。研究者通过DENV感染试验, 从180万种化合物中筛选出了一种新型抗登革病毒化合物(NITD-982, 22), 它能抑制宿主二氢乳酸脱氢酶(DHODH), 而DHODH是嘧啶生物合成所必需的酶。NITD-982就是通过消耗细胞内嘧啶来抑制病毒RNA合成的(EC50 = 2.4 µmol·L-1, CC50 > 5 µmol·L-1)。与体外药效相比, 该化合物在DENV-AG129小鼠模型中没有显示出任何效果。体内疗效的缺乏可能是由于从饮食中外源性摄取嘧啶, 或者是由于当前化合物具有较高的血浆蛋白结合活性。尽管如此, NITD-982仍不失为一种潜在的抗病毒先导化合物[46]。
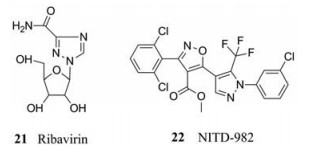
|
Baltina等[47]通过DENV-2型在非洲绿猴肾细胞中的细胞病变效应(CPE)和病毒感染性的抑制试验, 阐明了甘草酸(GL)衍生物的结构-抗病毒活性关系, 同时证明了GL (96%纯度)对非洲绿猴肾细胞具有较低的细胞毒性, 它可以抑制DENV-2诱导的CPE以及降低DENV-2的感染性(EC50 = 8.1 µmol·L-1)。因此, 对GL的结构进行修饰可能是寻找抗DENV-2感染药物的新方法。GL及其衍生物如图 6所示。

|
Figure 6 The structure of GL and its derivatives (compound 23-34)[47]. (Figure derived from Baltina LA, et al. Bioorg Med Chem Lett, 2019: 126645) |
近年来, DENV的生物学和抗病毒药物研究取得了很大的进展, 获得了一些有效的抗DENV候选药物。但DENV药物研发仍然面临诸多挑战:首先, 缺少对4种血清型登革病毒都有效的抗登革病毒药物; 其次, 重症登革病毒感染的分子机制目前还不清楚, 这在很大程度上限制了抗登革病毒药物的研发; 再次, 目前还没有合适的能够进行药物体内效果研究的小动物模型等, 这些都是亟需解决的重要问题。
现有DENV抑制剂的研发策略主要有3种: ①基于酶靶标的高通量筛选, 病毒多聚酶和蛋白酶是抗病毒药物研发中的重要靶标, 通过基于病毒多聚酶和蛋白酶的高通量筛选, 人们获得了一些活性化合物。②基于表型的高通量筛选, 由于病毒感染的过程复杂, 导致很多酶水平表现出高活性的化合物在进一步细胞实验或动物实验中没有理想的效果, 而基于表型的高通量筛选, 不仅能更加有效的寻找对病毒感染过程确切有效且结构新颖的化合物, 还有希望获得新的作用靶点。③从已知的HCV抑制剂中寻找登革病毒抑制剂, 由于登革病毒和HCV同属黄病毒, 结构上有很大的相似性, 因此从已知的HCV抑制剂出发, 有希望获得有效的登革病毒抑制剂。
现有的登革病毒抑制剂也依然存在一些缺点, 例如蛋白酶虽然在维持病毒的活性方面也起着关键作用, 但蛋白酶抑制剂容易诱导耐药性病毒株的产生; 很多病毒多聚酶抑制剂活性不够高以及存在很多毒副作用; 病毒侵入抑制剂尽管在体外能够表现出一定的抗登革病毒活性, 但缺乏一些有效的体内抗病毒效果评估或者体内抗病毒效果不佳, 很难应用于登革病毒感染的临床治疗等。总而言之, 发现新一代登革病毒抑制剂依然是未来抗病毒药物的研究热点。
| [1] |
Martina BE, Koraka P, Osterhaus AD. Dengue virus pathogenesis: an integrated view[J]. Clin Microbiol Rev, 2009, 22: 564-581. DOI:10.1128/CMR.00035-09 |
| [2] |
Uno N, Ross TM. Dengue virus and the host innate immune response[J]. Emerg Microbes Infect, 2018, 7: 1-11. |
| [3] |
Green J, Bandarage U, Luisi K, et al. Recent advances in the discovery of dengue virus inhibitors[J]. Annu Rep Med Chem, 2012, 47: 297-317. |
| [4] |
Muller DA, Young PR. The flavivirus NS1 protein: molecular and structural biology, immunology, role in pathogenesis and application as a diagnostic biomarker[J]. Antiviral Res, 2013, 98: 192-208. |
| [5] |
Dighe SN, Dua K, Chellappan DK, et al. Recent update on anti-dengue drug discovery[J]. Eur J Med Chem, 2019, 176: 431-455. DOI:10.1016/j.ejmech.2019.05.010 |
| [6] |
Yauch LE, Shresta S. Dengue virus vaccine development[J]. Adv Virus Res, 2014, 88: 315-372. DOI:10.1016/B978-0-12-800098-4.00007-6 |
| [7] |
Chen YL, Yin Z, Duraiswamy J, et al. Inhibition of dengue virus RNA synthesis by an adenosine nucleoside[J]. Antimicrob Agents Chemother, 2010, 54: 2932-2939. DOI:10.1128/AAC.00140-10 |
| [8] |
Schul W, Liu W, Xu HY, et al. A dengue fever viremia model in mice shows reduction in viral replication and suppression of the inflammatory response after treatment with antiviral drugs[J]. J Infect Dis, 2007, 195: 665-674. DOI:10.1086/511310 |
| [9] |
Yin Z, Chen YL, Schul W, et al. An adenosine nucleoside inhibitor of Dengue virus[J]. Proc Natl Acad Sci U S A, 2009, 106: 20435-20439. DOI:10.1073/pnas.0907010106 |
| [10] |
Yin Z, Chen YL, Kondreddi RR, et al. N-Sulfonylanthranilic acid derivatives as allosteric inhibitors of Dengue viral RNA-dependent RNA polymerase[J]. J Med Chem, 2009, 52: 7934-7937. DOI:10.1021/jm901044z |
| [11] |
Niyomrattanakit P, Chen YL, Dong H, et al. Inhibition of dengue virus polymerase by blocking of the RNA tunnel[J]. J Virol, 2010, 84: 5678-5686. DOI:10.1128/JVI.02451-09 |
| [12] |
Yang CC, Hsieh YC, Lee SJ, et al. Novel dengue virus-specific NS2B/NS3 protease inhibitor, BP2109, discovered by a high-throughput screening assay[J]. Antimicrob Agents Chemother, 2011, 55: 229-238. |
| [13] |
Lin C, Yu J, Hussain M, et al. Design, synthesis, and biological evaluation of novel 7-deazapurine nucleoside derivatives as potential anti-dengue virus agents[J]. Antiviral Res, 2018, 149: 95-105. DOI:10.1016/j.antiviral.2017.11.005 |
| [14] |
Migliaccio G, Tomassini JE, Carroll SS, et al. Characterization of resistance to non-obligate chain-terminating ribonucleoside analogs that inhibit hepatitis C virus replication in vitro[J]. J Biol Chem, 2003, 278: 49164-49170. DOI:10.1074/jbc.M305041200 |
| [15] |
Olsen DB, Eldrup AB, Bartholomew L, et al. A 7-deaza-adenosine analog is a potent and selective inhibitor of hepatitis C virus replication with excellent pharmacokinetic properties[J]. Antimicrob Agents Chemother, 2004, 48: 3944-3953. DOI:10.1128/AAC.48.10.3944-3953.2004 |
| [16] |
Chen YL, Yin Z, Lakshminarayana SB, et al. Inhibition of Dengue virus by an ester prodrug of an adenosine analog[J]. Antimicrob Agents Chemother, 2010, 54: 3255-3261. DOI:10.1128/AAC.00397-10 |
| [17] |
Chen YL, Yokokawa F, Shi PY. The search for nucleoside/nucleotide analog inhibitors of Dengue virus[J]. Antiviral Res, 2015, 122: 12-19. DOI:10.1016/j.antiviral.2015.07.010 |
| [18] |
Timiri AK, Sinha BN, Jayaprakash V. Progress and prospects on DENV protease inhibitors[J]. Eur J Med Chem, 2016, 117: 125-143. DOI:10.1016/j.ejmech.2016.04.008 |
| [19] |
De Clercq E. Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV[J]. Int J Antimicrob Agents, 2009, 33: 307-320. DOI:10.1016/j.ijantimicag.2008.10.010 |
| [20] |
Wyles DL. Antiviral resistance and the future landscape of hepatitis C virus infection therapy[J]. J Infect Dis, 2013, 207: S33-S39. DOI:10.1093/infdis/jis761 |
| [21] |
Nitsche C, Behnam MA, Steuer C, et al. Retro peptide-hybrids as selective inhibitors of the Dengue virus NS2B-NS3 protease[J]. Antiviral Res, 2012, 94: 72-79. DOI:10.1016/j.antiviral.2012.02.008 |
| [22] |
Pambudi S, Kawashita N, Phanthanawiboon S, et al. A small compound targeting the interaction between nonstructural proteins 2B and 3 inhibits Dengue virus replication[J]. Biochem Biophys Res Commun, 2013, 440: 393-398. DOI:10.1016/j.bbrc.2013.09.078 |
| [23] |
Oliveira AS, Silva ML, Oliveira AF, et al. NS3 and NS5 proteins: important targets for anti-dengue drug design[J]. J Braz Chem Soc, 2014, 25: 1759-1769. |
| [24] |
Byrd CM, Grosenbach DW, Berhanu A, et al. Novel benzoxazole inhibitor of Dengue virus replication that targets the NS3 helicase[J]. Antimicrob Agents Chemother, 2013, 57: 1902-1912. DOI:10.1128/AAC.02251-12 |
| [25] |
Aguilera-Pesantes D, Robayo LE, Méndez PE, et al. Discovering key residues of Dengue virus NS2b-NS3-protease: new binding sites for antiviral inhibitors design[J]. Biochem Biophys Res Commun, 2017, 492: 631-642. DOI:10.1016/j.bbrc.2017.03.107 |
| [26] |
Leonel CA, Lima WG, Dos Santos M, et al. Pharmacophoric characteristics of Dengue virus NS2B/NS3pro inhibitors: a systematic review of the most promising compounds[J]. Arch Virol, 2018, 163: 575-586. |
| [27] |
Lee JC, Chang FR, Chen SR, et al. Anti-dengue virus constituents from formosan zoanthid Palythoa mutuki[J]. Mar Drugs, 2016, 14: 151. DOI:10.3390/md14080151 |
| [28] |
de Sousa LR, Wu H, Nebo L, et al. Flavonoids as noncompetitive inhibitors of Dengue virus NS2B-NS3 protease: inhibition kinetics and docking studies[J]. Bioorg Med Chem, 2015, 23: 466-470. DOI:10.1016/j.bmc.2014.12.015 |
| [29] |
Li L, Basavannacharya C, Chan KW, et al. Structure-guided discovery of a novel non-peptide inhibitor of Dengue virus NS2B-NS3 protease[J]. Chem Biol Drug Des, 2015, 86: 255-264. |
| [30] |
Miller S, Sparacio S, Bartenschlager R. Subcellular localization and membrane topology of the Dengue virus type 2 non-structural protein 4B[J]. J Biol Chem, 2006, 281: 8854-8863. DOI:10.1074/jbc.M512697200 |
| [31] |
Xie X, Zou J, Wang QY, et al. Targeting dengue virus NS4B protein for drug discovery[J]. Antiviral Res, 2015, 118: 39-45. DOI:10.1016/j.antiviral.2015.03.007 |
| [32] |
Xie X, Wang QY, Xu HY, et al. Inhibition of dengue virus by targeting viral NS4B protein[J]. J Virol, 2011, 85: 11183-11195. DOI:10.1128/JVI.05468-11 |
| [33] |
Alen MM, Schols D. Dengue virus entry as target for antiviral therapy[J]. J Trop Med, 2012, 2012: 628475. |
| [34] |
De La Guardia C, Lleonart R. Progress in the identification of dengue virus entry/fusion inhibitors[J]. Biomed Res Int, 2014, 2014: 825039. |
| [35] |
Hidari K, Abe T, Suzuki T. Carbohydrate-related inhibitors of dengue virus entry[J]. Viruses, 2013, 5: 605-618. DOI:10.3390/v5020605 |
| [36] |
Costin JM, Jenwitheesuk E, Lok SM, et al. Structural optimization and de novo design of dengue virus entry inhibitory peptides[J]. PLoS Negl Trop Dis, 2010, 4: e721. DOI:10.1371/journal.pntd.0000721 |
| [37] |
Lok SM, Costin JM, Hrobowski YM, et al. Release of dengue virus genome induced by a peptide inhibitor[J]. PLoS One, 2012, 7: e50995. |
| [38] |
Modis Y, Ogata S, Clements D, et al. A ligand-binding pocket in the dengue virus envelope glycoprotein[J]. Proc Natl Acad Sci U S A, 2003, 100: 6986-6991. DOI:10.1073/pnas.0832193100 |
| [39] |
Jadav SS, Kaptein S, Timiri A, et al. Design, synthesis, optimization and antiviral activity of a class of hybrid dengue virus E protein inhibitors[J]. Bioorg Med Chem Lett, 2015, 25: 1747-1752. DOI:10.1016/j.bmcl.2015.02.059 |
| [40] |
Behnam MA, Nitsche C, Boldescu V, et al. The medicinal chemistry of dengue virus[J]. J Med Chem, 2016, 59: 5622-5649. DOI:10.1021/acs.jmedchem.5b01653 |
| [41] |
Ma L, Jones CT, Groesch TD, et al. Solution structure of dengue virus capsid protein reveals another fold[J]. Proc Natl Acad Sci U S A, 2004, 101: 3414-3419. DOI:10.1073/pnas.0305892101 |
| [42] |
Byrd CM, Dai D, Grosenbach DW, et al. A novel inhibitor of dengue virus replication that targets the capsid protein[J]. Antimicrob Agents Chemother, 2013, 57: 15-25. DOI:10.1128/AAC.01429-12 |
| [43] |
Scaturro P, Trist IM, Paul D, et al. Characterization of the mode of action of a potent dengue virus capsid inhibitor[J]. J Virol, 2014, 88: 11540-11555. DOI:10.1128/JVI.01745-14 |
| [44] |
Pastorino B, Nougairède A, Wurtz N, et al. Role of host cell factors in flavivirus infection: implications for pathogenesis and development of antiviral drugs[J]. Antiviral Res, 2010, 87: 281-294. DOI:10.1016/j.antiviral.2010.04.014 |
| [45] |
Rodenhuis-Zybert IA, Wilschut J, Smit JM. Dengue virus life cycle: viral and host factors modulating infectivity[J]. Cell Mol Life Sci, 2010, 67: 2773-2786. |
| [46] |
Wang QY, Bushell S, Qing M, et al. Inhibition of Dengue virus through suppression of host pyrimidine biosynthesis[J]. J Virol, 2011, 85: 6548-6556. DOI:10.1128/JVI.02510-10 |
| [47] |
Baltina LA, Tasi YT, Huang SH, et al. Glycyrrhizic acid derivatives as dengue virus inhibitors[J]. Bioorg Med Chem Lett, 2019, 29: 126645. DOI:10.1016/j.bmcl.2019.126645 |