 2020, Vol. 55
2020, Vol. 55



丙型肝炎病毒(hepatitis C virus, HCV)是一种血液传播性疾病, 通常会被描述为“沉默杀手”, 主要是因为丙肝病毒的潜伏期很长, 而且极容易慢性化。慢性丙肝会对肝脏造成不同程度的损伤, 如不及时治疗, 会引起一系列的并发症, 如肝纤维化、肝硬化甚至是肝癌。估计全球仍有1.3亿~1.85亿人感染HCV, 每年大约有70万人死于丙型肝炎及其并发症。我国的丙肝病毒感染者大约有1 000万人[1]。
HCV病毒进入人体, 会选择性感染肝脏细胞, 利用肝细胞中的物质进行增殖。其生命周期主要分为吸附与融合、翻译和RNA复制、组装、出芽和释放(图 1)[2]。抗HCV药物通过作用于病毒编码蛋白或致病相关宿主细胞因子, 从不同阶段阻断HCV在肝细胞中的生命周期, 从而达到抗病毒疗效。根据作用靶点的不同, 抗HCV药物主要包括直接抗病毒药物(direct-acting antiviral agents, DAA)和宿主靶向药物(host-targeting agents, HTA)。其中, DAA直接作用于病毒蛋白, 往往具有较高的应答率和治愈率[3]。本综述精选近几年研究实例, 从药物化学的角度总结了HCV治疗历程及DAA研究的最近进展。

|
Figure 1 The life cycle of hepatitis C virus (HCV) |
1989年, 科学家首次发现丙型肝炎病毒。1991年注射用干扰素α (IFNα)被美国FDA批准用于HCV治疗, 从此开始了丙肝治疗的干扰素时代, 当时的治愈率仅为6%。2001年各国一致推荐的标准治疗方案:长效干扰素与利巴韦林联合使用, 可获得中等的持续病毒学应答(sustained viral response, SVR), 治愈率进一步提升到41%, 但这一标准治疗方案存在治愈率较低、患者耐受性差、治疗周期长(48周~72周)等问题[4]。2011年, FDA批准的特拉匹韦(telaprevir)和波普瑞韦(boceprevir)开启了丙肝DAA治疗的新时代, 结束了干扰素时代。之后, 利巴韦林(ribavirin, RBV)、聚乙二醇化长效干扰素α (IFN-α 2a或IFN-α 2b)和telaprevir或boceprevir组成的三联疗法大幅提高了治愈率, 也显著地缩短了治疗周期(24周~48周), 使得HCV-1型初治患者的SVR提高至68%~75%。但是三联法具有长期毒副作用、高成本和较高的复发率等问题。2013年, NS5B聚合酶抑制剂索菲布韦(sofosbuvir)的上市, 使SVR提高至90%以上, 基本可以达到临床治愈, 且不良反应少, 疗程较短。但是单一DAA治疗的疗效总是不足, 并且容易出现耐药毒株。需要不同机制的DAA (NS3/4A、NS5A、NS5B抑制剂)联合治疗以降低耐药毒株的发生率和剂量相关的毒性以及增强治疗的有效性。因此, 基于几种不同作用方式的DAA的组合, 是目前治疗HCV感染最重要的需求。
目前, 欧洲药物管理局(EMA)和美国食品药品管理局(FDA)批准用于HCV治疗的DAA药物[5]如表 1所示, 基于DAA药物组合疗法[6]如图 2所示。
| Table 1 HCV inhibitor targets and corresponding drugs |

|
Figure 2 The direct-acting antiviral agents (DAA)-based combination therapies |
NS3/4A蛋白是一种多功能酶, 具有N端丝氨酸蛋白酶结构域和C端解旋酶结构域, 主要功能为催化裂解HCV多聚蛋白前体, 释放出成熟的丝氨酸蛋白酶、螺旋酶/核苷酸三磷酸酶和RNA依赖性RNA聚合酶(RNA dependent RNA polymerase, RdRp)。蛋白酶抑制剂(protease inhibitor, PI)通过与NS3/4A蛋白酶活性中心发生可逆共价或非共价结合, 竞争性地抑制酶活性来阻断HCV复制、翻译和翻译后多聚蛋白的加工成熟; 此外PI还可以通过恢复感染细胞内的Ⅰ型干扰素信号通路, 提高机体的固有免疫[7]。但PI存在耐药位点多、不良反应发生率高、易受基因型影响等问题[8]。蛋白酶抑制剂可分为开环和大环两种。
1.1 开环类NS3/4A蛋白酶抑制剂通过消除格佐匹韦(grazoprevir, 1)中的P2-P4大环连接链, Rusere等[9]设计、合成了一系列开环类NS3/4A蛋白酶抑制剂, 代表化合物为2。开环化合物显示出对A156T突变株更好的抗病毒活性, 然而可能是由于构象灵活性的增加和与蛋白酶结合的相关熵补偿, 化合物对野生型(wild type, WT)蛋白酶和复制子(replicon)的活性降低。开环类抑制剂虽然通常比相应的大环类似物活性低, 但是相对更容易合成并且不容易出现耐药问题(图 3)。

|
Figure 3 The structures of compounds 1 and 2 |
化合物4是NS3蛋白酶抑制剂早期的先导化合物, 这种三肽酰基磺酰胺对基因1b型(genotype 1b, GT-1b)复制子细胞抑制活性比相应的羧酸(3)高100倍以上, 表明环丙基酰基磺酰胺部分对NS3/4A蛋白酶抑制活性的重要性, 但该化合物的药代动力学(pharmacokinetic, PK)性质很差, 通过继续优化发现了BMS-605339 (5), 但是由于该化合物的心血管风险而停止了临床前研究[10]。之后, 对BMS-605339的P2-异喹啉部分详细构效关系(structure-activity relationship, SAR)研究发现了阿那匹韦(asunaprevir, 6) (图 4), 其在兔心脏模型中没有心血管效应, 同时保持良好的活性(GT-1a和1b复制子的EC50 < 10 nmol·L-1)和PK性质[11], 随后进入临床开发, 并在日本被批准为SUNVEPRATM, 与PEG-IFN/RBV联合使用。

|
Figure 4 The structures of compounds 3-6 |
基于吡嗪酮的HCV NS3蛋白酶抑制剂的设计源于化合物7[12], 用吡嗪酮代替7的P3部分, 得到等效抑制剂8。随后, 通过将不稳定的氨基甲酸酯基团替换为脲基团使抑酶活性增强10倍, 代表化合物为9。进一步延长化合物9的脲端取代基[13], 得到的化合物改善了对于NS3 GT-1a野生株和突变株活性。其中, 化合物10 (图 5)对GT-1a的野生株和耐药株R155K以及对GT-3a均显示出纳摩尔水平的抑制活性。

|
Figure 5 The structures of compounds 7-10 |
Zheng等[14]通过在P1环丙基氨基酸的环丙基上引入二氟甲基, 设计、合成了一系列三肽酰基磺酰胺类抑制剂, SAR研究表明:与CH3和CF3类似物相比, 该类衍生物抑酶活性分别增强了13倍和17倍。其中, 化合物11与NS3/4A蛋白酶共晶结构表明CHF2基团的C-H和Leu135主链羰基之间存在氢键作用。虽然该系列的PK性质较差, 但结果证明了在该氨基酸部分引入CHF2基团作为氢键供体有利于提高活性。
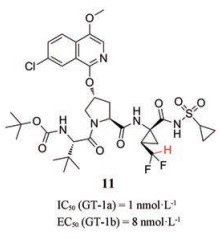
|
虽然早期的蛋白酶抑制剂对GT-1患者非常有效, 但对GT-3a及1b的多个耐药株的活性有待提高。早期研究发现修饰P2喹啉区域可以改善GT-3a的活性[15], Merck团队将化合物12的P2喹啉与脯氨酸部分形成螺环得到化合物13[16], 化合物13对GT-1b、3a的活性提高了约80倍, 并且提高了对GT-1b突变株的活性。基于化合物13, 通过P1-P3环化策略得到的化合物14, 进一步提高了对GT-1b、3a以及1b突变株的活性, 然而发现其PK性质差(AUC0-24 h = 0.14 μmol·L-1·h; t1/2 = 0.9 h; F = 4%)。在已有构效关系的基础上, 对化合物14的P2芳香族区域和P4氨基甲酸酯部位进行优化, 发现化合物MK-8831 (15) (图 6), PK性质得到改善(F > 17%), 其对GT-1的IC50值达到皮摩尔级(GT-1a和1b, IC50 = 9和4 pmol·L-1), 抗病毒活性较好(GT1-3, EC50 = 0.7~3.4 nmol·L-1)。

|
Figure 6 The structures of compounds 12-15 |
之后, Merck团队又通过将螺环脯氨酸结构引入MK-5172 (grazoprevir, 1)的骨架结构得到一系列衍生物[17], 在抑酶和细胞活性测试中, 化合物16和17对不同的基因型和突变株均显示出优异的活性。以及在螺环化合物基础上, 增加P1-P3环化得到的双大环18也显示出优异的抑酶和抗病毒活性(图 7)。

|
Figure 7 The structures of compounds 16-18 |
所有NS3/4A PI都易受耐药性的影响, 值得注意的是, 几乎所有PI治疗失败的患者都存在D168A/V突变[18]。野生型和突变蛋白酶与抑制剂的高分辨率的共晶结构确定了单点突变导致的耐药性的分子机制。这些晶体结构揭示了PI的大杂环P2部分结合在底物结合区(被定义为底物包膜)以外, 并与残基Arg155、Ala156和Asp168进行广泛的相互作用。突变导致的Arg155和Asp168之间的静电相互作用的破坏是对NS3/4A PI产生耐药的原因。Merck团队运用底物包膜假说[19], 设计并合成了一系列P1-P3大环类衍生物(图 8), 发现在P2喹喔啉部分的3-位具有小疏水性取代基的抑制剂对耐药株具有很好的活性, 抑制主要耐药突变株(EC50 ≤ 5 nmol·L-1)。这些发现支持这一观点:设计与蛋白酶包膜内残基相互作用, 同时避免与对底物识别不重要的残基相互作用的抑制剂可以提高抗耐药性。

|
Figure 8 The structures of P1-P3 macrocyclic derivatives |
吉利德公司选用次膦酸等排体代替P1-羧酸, 设计合成了一系列P-苄基取代的衍生物[20], 其中2, 6-二氟苄基取代衍生物GS-9256 (19), 在GT-1b Huh-luc细胞测试中表现出较好的抑制活性(EC50 = 20 nmol·L-1)和较小的细胞毒性[21]。最近的研究发现GS-9256与NS5A抑制剂ledipasvir以及其他药物(包括干扰素-α、利巴韦林、NS5B聚合酶抑制剂GS-6620和tegobuvir)组合使用具有协同作用。此外, GS-9256在小鼠中具有高生物利用度(接近100%), 可进一步应用于治疗GT-1患者的慢性HCV感染的临床开发。
Bowsher等[22]设计、合成了萘连接的P2-P4大环酰基磺酰胺类衍生物, 在个位数纳摩尔级别抑制基因1a和1b型的HCV NS3蛋白酶。通过对大环连接链以及P1亚位点的结构修饰来优化该系列中化合物的PK性质。最终得到大鼠口服生物利用度为100%、血浆半衰期长的化合物20。然而, 该化合物在离体兔心脏模型中表现出心脏毒性而停止了进一步的优化工作。
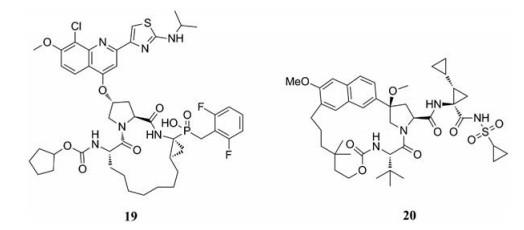
|
NS5B是RNA依赖性RNA聚合酶, 在病毒复制周期中, 以病毒RNA链为模板, 通过催化核糖核苷三磷酸的聚合来完成HCV RNA的复制过程。因此靶向NS5B聚合酶可以直接抑制病毒RNA合成, 阻断病毒的复制周期。在结构上, RdRp具有聚合酶典型的“右手”结构, 包括拇指(thumb)、手指(fingers)、和手掌(palm) 3个结构域[23]。手掌区的功能是形成核苷酸转移反应的催化中心; 手指区主要是与复制所需的三磷酸核苷酸相互作用; 而拇指区则是在RNA复制起始和延伸过程中发挥作用。NS5B聚合酶还包括5个变构位点(Thumb 1、Thumb Ⅱ、Palm Ⅰ、Palm Ⅱ、Palm Ⅲ), 调节聚合酶的基本构象来保持催化活性。
人体细胞中不含有与NS5B RdRp功能相近的酶, 这就使得NS5B RdRp的抑制剂具有高度的选择性, 因此NS5B聚合酶成为一个理想的抗病毒药物靶点。根据其结构不同分为核苷类和非核苷类聚合酶抑制剂两大类, 这两类药物作用机制不同, 可以联合应用。
2.1 核苷类NS5B聚合酶抑制剂核苷类聚合酶抑制剂(nucleoside analogue polymerase inhibitor, NPI)是经过糖基化或碱基化修饰的核苷类似物, 通过模拟酶的天然底物, 竞争作用于NS5B的催化活性位点, 插入到新合成的核苷酸链中, 使链的延伸终止, 从而阻断HCV的生命周期。NS5B的活性位点具有高度保守性, 不易产生突变, 因此NPI具有泛基因型和高抗耐药性特征[24]。但是NPI需要在体内转化为活性三磷酸盐的形式才能发挥抑制聚合酶活性, 且第一步5'-单磷酸核苷的形成是磷酸化过程的限速步骤, 为了改善NPI的膜渗透性、提高NPI在肝细胞内的三磷酸盐水平, 多数采用前药策略。
索菲布韦(sofosbuvir, 21) (图 9)是一种最初由Pharmasset开发的氨基磷酸酯前药, 是迄今为止唯一一个获得HCV治疗批准的核苷酸前药, 其显示出高的抗耐药性和泛基因型活性。此药联合NS5A蛋白酶抑制剂ledipasvir可治愈慢性丙型肝炎(chronic hepatitis C, CHC), 这对CHC治疗具有里程碑意义[3]。然而, 该抑制剂不可避免地在NS5B中诱导S282T突变, 该突变显著降低了活性三磷酸盐代谢物与NS5B结合的亲和力, 因此降低了sofosbuvir的复制子活性, 此外, 在临床试验中也检测到S282T突变[25]。

|
Figure 9 The structures of compounds 21-25 |
由于发现HCV候选药物mericitabine的活性代谢物(22)比sofosbuvir的尿苷代谢物能更有效地抑制野生型和S282T突变株的NS5B聚合酶。因此, Zhen等[26]基于化合物22, 应用双前药策略设计一系列具有N4修饰胞嘧啶的氨基磷酸酯衍生物。其中, 化合物23 (EC50 = 0.366 μmol·L-1)相比sofosbuvir (EC50 = 0.589 μmol·L-1)对S282T突变株HCV复制子表现出更强的活性。此外, 这些化合物在高达100 μmol·L-1浓度下不具有细胞毒性(图 9)。
2017年, Zhou等[27]用氯原子取代sofosbuvir中2'-甲基得到化合物24, 化合物24表现出较高的泛基因型活性(GT1-4, EC50 = 0.05~0.17 μmol·L-1), 其活性代谢物24-三磷酸(24-TP, 25)对多个基因型的野生株和S96T突变株具有微摩尔水平的抑制活性, 且具有较长的半衰期(t1/2 = 16 h)。化合物24在人血浆中的稳定性以及人肠微粒体和肝微粒体中的代谢稳定性, 使其作为候选药物得到进一步研究(图 9)。
Jonckers课题组[28]设计、合成了一类2'-脱氧-2'-螺氧杂环丁烷核糖核苷的氨基磷酸酯前药衍生物, 通过Huh7-复制子细胞活性测试, 发现大多数衍生物显示显著的活性(EC50 = 0.23~96.4 μmol·L-1)和低的细胞毒性(CC50 > 98.4 μmol·L-1)。
2016年, 该课题组又报道了2'-脱氧-2'-螺氧杂环丁烷尿苷核苷酸的环状磷酸酯衍生物JNJ-54257099 (26)[29], 尽管该化合物在体外基于Huh-7复制子细胞的活性测试中没有表现出任何抗HCV活性, 但安全性和PK特征较好。在人原代肝细胞中对化合物26的体外培养, 以及临床前其他物种的药代动力学研究, 共同揭示了2'-脱氧-2'-螺氧杂环丁烷尿苷三磷酸(27)高水平的形成。27是HCV NS5B聚合酶的有效抑制剂(IC50 = 1 μmol·L-1) (图 10)。特别是在建立的HCV感染(GT-1a和3a)的小鼠模型中, 口服给药26后观察到病毒HCV RNA的剂量依赖性减少。

|
Figure 10 The structures of compounds 26 and 27 |
非核苷类聚合酶抑制剂(non-NPI, NNPI)以非竞争方式与NS5B聚合酶催化部位的变构位点结合, 在延伸复合物形成前, 导致多聚蛋白复制复合体的重要构象发生改变, 从而抑制NS5B聚合酶活性中心的催化效率, 干扰病毒的体内复制过程。与核苷类聚合酶抑制剂相比, 非核苷类聚合酶抑制剂基因屏障较低, 容易发生耐药, 对HCV基因型也有选择性。这类NS5B抑制剂可能需要联合作用于其他靶点的抗HCV药物解决这些问题。该类抑制剂因结构不同又分为喹诺酮类、噻吩羧酸类、苯并呋喃酮等多种骨架抑制剂。
2.2.1 喹诺酮类衍生物为了提高2-芳基喹诺酮类非核苷类NS5B聚合酶抑制剂28和29的抗病毒活性, Cheng等[30]通过分子杂合策略发现了2-烷基-N-苄基喹诺酮类衍生物, 该类化合物的抗病毒活性提高, 尤其以化合物30活性最好(GT-2a Huh7.5.1细胞, EC50 = 0.4 μmol·L-1)。但是这些多取代的喹诺酮具有不良药代动力学性质, 鉴于此, Cheng等又通过骨架跃迁策略设计、合成一系列1, 6-二氮杂萘-4, 5-二酮类衍生物以改善化合物的类药性, 代表化合物为31 (EC50 = 2.5 μmol·L-1) (图 11)。分子模拟表明: 2-烷基喹诺酮类和1, 6-二氮杂萘-4, 5-二酮类衍生物均与HCV NS5B聚合酶的拇指Ⅱ区(Thumb Ⅱ)结合而发挥作用。

|
Figure 11 The structures of compounds 28-31 |
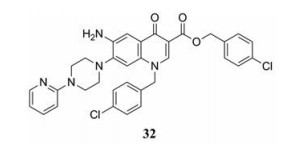
2015年, Manfroni等[31]设计、合成了一系列6氨基-7-[4-(2-吡啶基)-1-哌嗪基]喹诺酮类衍生物, 其中化合物32的活性与选择性最好, 抑制NS5B聚合酶的IC50值为0.069 μmol·L-1, 在GT-1b细胞中抑制HCV复制的EC50值为3.03 μmol·L-1, 细胞毒性较低(CC50 > 163 μmol·L-1)。
2.2.2 噻吩羧酸类Lomibuvir (33)是结合HCV NS5B聚合酶Thumb Ⅱ的非核苷类抑制剂, 为了改善其抗病毒活性和PK性质, Court等[32]用甘氨酸衍生的酰胺取代33中的反式-4-羟基环己烷环, 得到与化合物33活性相当、物理化学性质改善的一系列化合物, 代表化合物为34 (图 12)。

|
Figure 12 Model of 33 bound to HCV genotype 1b NS5B and derivatives (PDB: 2GIR) |
化合物33与NS5B聚合酶的晶体结构显示:化合物33的反式-4-羟基环己基靠近形成结合口袋侧面的残基Arg501。2017年, Li等[33]运用基于结构的药物设计和构象限制策略, 设计、合成一系列内酰胺类抑制剂, 预期与Arg501残基产生额外的相互作用。发现化合物35相对化合物33的抑酶活性和抗病毒活性均提高了3~5倍, 验证了设计思想。此外, 化合物35具有类药性和与进一步开发所需参数一致的体内特征(图 12)。
|
噻吩羧酸类化合物36是与NS5B蛋白酶Thumb Ⅱ结合的抑制剂[34]。用内酰胺代替化合物36中酰胺使抑酶活性得到改善, 尤其是对复制子细胞的抑制活性得到很大的提升, 代表化合物为37。为了减少化合物37对孕烷X受体(PXR)的活化, 进一步引入极性基团来破坏PXR配体结合口袋中疏水区域的相互作用, 发现化合物38不仅减少对PXR的活化而且保持细胞抑制活性, 同时该化合物还具有良好的PK性质(图 13)。

|
Figure 13 The structures of compounds 36-38 |
苯并呋喃酮类化合物39是靶向NS5B手掌区的变构抑制剂, 抑酶活性及细胞水平的抗病毒活性均较弱。BMS团队通过延长化合物39的C5位片段发现BMS-929075 (40)[35]。除基因2型外, 化合物40对所有基因型表现出有效的HCV抑制作用, 且化合物40在3种临床前动物模型中表现出一致的高口服生物利用度(F > 48%)。此外, 通过使用稳定的无定形固体分散剂解决了BCS II类化合物的溶解度限制吸收的问题, 在健康志愿者完成单次递增剂量(single ascend dose, SAD)和多次递增剂量(multiple ascend dose, MAD) Ⅰ期临床试验中发现40的PK性质优于预期, 表明化合物40具有一天给药一次的潜力。
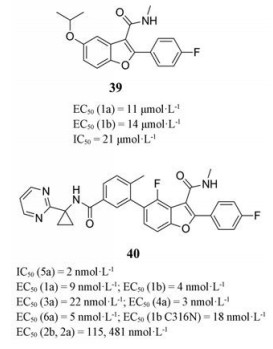
|
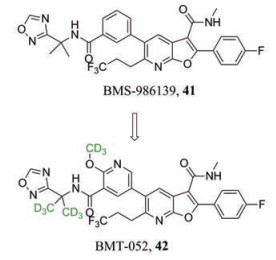
由于第二代泛基因型抑制剂BMS-986139[36] (41)在毒理学研究中出现的微晶化问题, 研究人员停止了对其进一步的研究。之后, BMS团队通过在BMS-986139的C5苯环和酰胺取代基中添加氘原子发现BMT-052 (42)[37], 化合物42是有效的泛基因型HCV抑制剂, 对基因1-6型以及1b C316N突变株的EC50值为1~7 nmol·L-1, 在3种临床前物种中表现出良好口服生物利用度(F > 85%)。
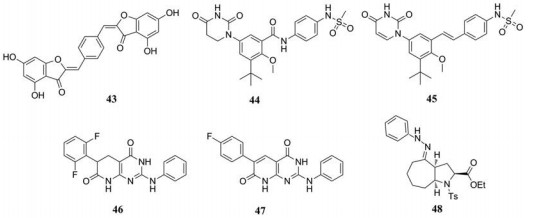
2-亚苄基苯并呋喃-3(2H)-酮是强效的黄酮类HCV小分子抑制剂, 该类衍生物的结合位点位于拇指Ⅰ区(Thumb Ⅰ)。由于发现Thumb Ⅰ具有未开发的疏水区域, 可容纳更大的结构。Meguellati等[38]假设二聚体橙酮比单体衍生物能更好的结合Thumb Ⅰ, 将会产生更高的抑制活性。因此, 设计合成一系列连接链不同的二聚体橙酮作为NS5B聚合酶抑制剂的新型骨架。通过NS5B酶活测试, 发现大多数衍生物抑酶水平在微摩尔级别, 其中, 化合物43抑酶活性最高(IC50 = 1.3 μmol·L-1)。但是分子模拟表明43的结合位点可能是Palm Ⅰ, 而不是单体衍生物结合的Thumb Ⅰ。
|
二氢尿嘧啶类似物44是一种有效的GT-1a和1b聚合酶抑制剂, EC50值分别为51和19 nmol·L-1, 然而, 化合物44的血浆清除率高和口服生物利用度低(F = 1.4%)。用反式烯烃取代44中的酰胺键能改善化合物的PK性质, 在此基础上, AbbVie团队继续用N-连接的尿嘧啶代替二氢尿嘧啶得到ABT-072 (45), 其对GT-1a和1b的EC50值分别为1和0.3 nmol·L-1, 且血浆半衰期延长, 口服生物利用度提高到44%。ABT-072与ritonavir和利巴韦林联合治疗基因1型患者的Ⅱ期临床研究中, 91%的患者在治疗24周后获得持续病毒学应答(SVR24), 证明了ABT-072联合用药治疗基因1型HCV患者的有效性[39]。
Camarasa等[40]利用吉利德公司报道的一系列HCV抑制剂—吡啶并[3, 2-d]嘧啶的结构信息, 设计合成了吡啶并[2, 3-d]嘧啶-7(8H)-酮化合物。通过含有HCV亚型1b复制子的稳定细胞系测定EC50, 发现46和47表现出很好的活性(EC50值分别为0.027和0.034 μmol·L-1)和高选择性(SI = 196、397)。分子模拟揭示46和47通过与变构位点结合抑制NS5B聚合酶。
Kaushik-Basu等[41]报道的双环八氢环庚并[b]吡咯-4(1H)-酮类衍生物对HCV基因1b和2a型复制子具有抑制活性。化合物48是其中最有效的衍生物, 对基因1b和2a型的EC50值分别为1.8和4.5 μmol·L-1, 并且毒性很小(CC50 > 200 μmol·L-1)。初步的构效关系研究证实这类衍生物的酯功能与顺式稠合环结合是获得基因1b和2a型抗病毒活性的基本特征。
3 NS5A抑制剂HCV NS5A蛋白是由447个氨基酸组成的高度磷酸化蛋白, 包括3个结构域。其中, 结构域1对HCV基因组的复制是必不可少的, 是药物设计的关注重点; 结构域2和3在RNA复制和病毒颗粒组装环节中发挥重要作用。
|
NS5A上丝氨酸残基产生两种磷酸化程度不同的蛋白, 即基础磷酸化p56和高度磷酸化p58, 均在HCV的生命周期中发挥重要作用。研究发现, NS5A可与HCV RNA、NS5B等组成复制复合体, 完成HCV复制; NS5A亦可通过与NS5B作用刺激NS5B合成负链RNA。此外, NS5A可通过其上的IFN敏感决定区与IFN刺激产生RNA依赖型蛋白激酶结合, 通过抑制其活性来调节对IFNα应答。NS5A功能的重要性和多样性使其成为抗HCV的重要靶点。且随着daclatasvir (DSV, 49)的发现, NS5A抑制剂吸引了更多的关注, 最显著的特征是这类抑制剂对HCV各个基因型均有较好的抗病毒效果[3]。
3.1 Declatasvir (DSV)类似物的研究DSV是由施贵宝研发的第一个对基因1-5型HCV均表现出显著抑制活性(EC50 = 9~146 pmol·L-1)的NS5A抑制剂[42], 于2015年获美国FDA批准上市。
目前, 研究人员已经开发了许多与DSV结构类似的NS5A抑制剂, 并且在体外显示出纳摩尔和皮摩尔抗HCV活性[43-49, 50]。这些抑制剂的结构可分为3个部分:两个对称或非对称的氨基酸或肽的基团(cap i、cap j)与不同类型和长度的中心结构域(core)。依据这个药效团模型, 以DSV作为对照, 根据结构变化的区域, 将本文的DSV类似物分为以下3类进行论述(图 14)。

|
Figure 14 The structure of declatasvir |
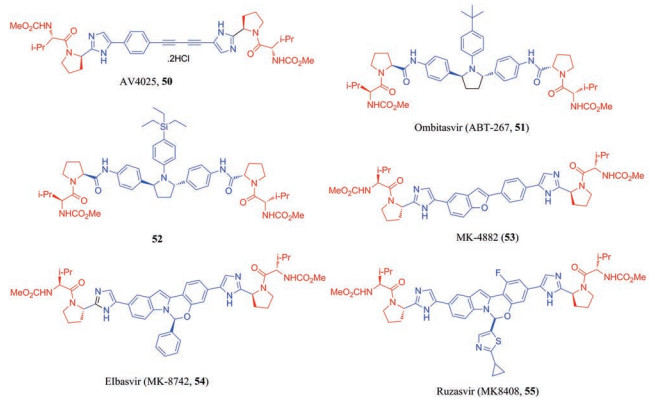
2014年, AllaChem公司使用二乙炔苯基结构代替DSV的中心区域得到的化合物AV4025 (50)[51], 其具有4个(S)手性中心, 改变任一个构型都能使活性降低。化合物50对GT-1a、2a和4a的EC50值分别为59、51和6.5 pmol·L-1, 此外, 化合物50在动物实验中表现出良好的PK性质, 且其在啮齿动物中耐受良好(在雄性小鼠中, LD50 ≥ 3435.4 ± 502.3 mg·kg-1)。化合物50对HCV感染的I/Ⅱ期临床试验结果乐观, 但未见后续报道。
AbbVie公司用反式-2, 5-二苯基-N-(4-叔丁基)苯基吡咯烷代替DSV中心区域得到ombitasvir (ABT-267, 51)。Ombitasvir对GT 1-5 HCV都显示出比DSV更强的体外抑制活性(EC50 = 1.7~19.3 pmol·L-1), 并且口服给药后在人体内半衰期长达约30 h[52]。2014年12月复方制剂Viekira Pa (ombitasvir/paritaprevir/ritonavir与dasabuvir的组合)被FDA批准上市, 并且可用于伴有肝硬化的患者。
将硅原子结合到药物分子中可以影响并且改善生物活性和ADMET性质[53-55], 其作为一种具有潜力的药物设计策略已引起关注。2018年, Liu等[56]基于ombitasvir, 采用C-Si转换策略得到一系列含硅衍生物。其中, 化合物52与ombitasvir相比, 对HCV GT 1-5的活性都有所增加(EC50 = 0.1~10 pmol·L-1), 特别是对GT-3a的抑制活性增加了7倍以上。此外, 化合物52在动物实验中表现出优异的PK性质和肝靶向性, 且在14天的重复剂量毒性研究中未观察到52的毒性。
Merck公司在MK-4882 (53)的基础上, 通过构象限制策略将苯并呋喃与苯环连成环, 并在环上引入苯基取代得到Elbasvir (MK-8742, 54), 54对GT1-4 HCV都有很好的抑制效果, 与已经上市的DSV和lediprasvir相比, 对多种耐药毒株的活性也表现更佳[57]。Elbasvir与grazoprevir组成的复方单片, 每日一次口服给药12周用于治疗HCV GT-1、4和6的感染者, GT-1、4和6的SVR12分别为94.4%、95.5%和96.4%[58]。这一复方单片于2016年1月被FDA批准上市用于治疗HCV GT-1和4的慢性感染。
|
2017年, Tong等[59]运用电子等排策略将elbasvir苯环替换为噻唑环衍生物得到MK-8408 (ruzasvir, 55), 其具有泛基因型活性, 对基因1-6型的EC90值为0.002~11 nmol·L-1, 且显示出对GT1a Y93H、L31V和GT2b (31M)等多个突变株活性改善, EC90值为0.002~0.067 nmol·L-1。在ruzasvir与uprifosbuvir联合用药的Ⅲ期临床试验中, ruzasvir 180 mg加uprifosbuvir 450 mg治疗HCV GT1、GT2、GT4、GT5和GT6感染者SVR12均高于90.9%, 且耐受性良好, 但对GT3感染者的疗效较低(SVR12 = 73.8%)[60]。
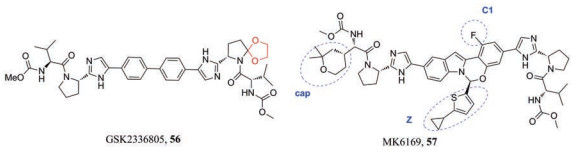
3.1.2 仅在两翼改变的类似物2014年, Kazmierski等[61]通过在DSV的一侧翼端的吡咯烷上引入氧杂螺环结构得到化合物GSK2336805 (JNJ56914845, 56), 化合物56对基因1a和1b型表现出皮摩尔抑制活性(EC50值分别为10.4和11.1 pmol·L-1), 对1b的野生株和L28V、L31V和Y93H突变株也显示出良好的活性(EC50 = 10.6~11.1 pmol·L-1)。JNJ56914845与simeprevir、ritonavir组成的3-DAA方案在治疗基因1a与1b型患者的Ⅱ期临床试验中, 分别有100%和93%的患者获得了持续病毒学应答(SVR12)。此外, Vijgen等[62]发现:在大多数患者中, 3-DAA组合治疗失败与所研究药物的耐药相关变异(resistance-associated variants, RAVs)的出现相关, 且在研究中观察到NS5A中的JNJ-56914845 RAV。
3.1.3 中心结构和两翼均改变的衍生物Yu等通过对elbasvir进行多样性修饰得到以下构效关系: ①在“帽” (cap)中引入四氢-2H-吡喃(THP)基团[63]可以改善对GT2b L31M的活性; ②四环吲哚核心的C-1位置为氟原子[64]可以改善对RAVs, 如GT1a Y93H和GT2b L31M的活性; ③ Z基团为4-环丙基苯基、4-二苯基[65]和7'-苯并二氢吡喃[66]等大芳香基团或者取代的噻唑和噻吩也提高对RAV的活性。Yu等将这些优势基团进行组合, 发现强效和泛基因型NS5A抑制剂MK-6169 (57), 对基因1a、1b、3a和4a型复制子的EC90值分别为4、4、1和6 nmol·L-1, 对GT1a Y93H和GT2b L31V的EC90分别为0.033和0.004 nmol·L-1, 同时该化合物还具有良好的PK特征。MK-6169作为临床前候选物得到进一步开发[67]。
Bae等[68]设计、合成了一系列具有联苯胺脯氨酰胺核心结构的抑制剂。构效关系显示:末端基团为苯基甘氨酸的抑制剂具有较高活性, 其中化合物BMK-20113 (58)活性最高, 对基因2a和1b型的EC50值分别为0.26和0.028 nmol·L-1。随后对化合物58进行心脏毒性、大鼠血浆稳定性和CYP450酶抑制活性研究, 表明化合物58是安全有效的(图 15)。
|

|
Figure 15 The structural evolution of compounds 58-61 |
2015年, Bae等[69]对化合物58进行新一轮修饰以提高其抗病毒活性, 用各种脯氨酸等排体代替L-脯氨酸, 联苯胺替换为取代的联苯胺衍生物得到一系列化合物。发现在联苯胺核心结构嵌入间位取代的抑制剂表现出最有效的抗HCV活性。其中, 化合物59不仅具有改善的抗病毒活性, 对基因2a和1b型的EC50值分别为0.059和0.003 nmol·L-1, 且对于真核细胞无毒并且通过Ames试验证明其不具有基因毒性。
2017年, You等[70]又设计将二芳基砜或硫酸盐嵌入到58的核心结构中并与咪唑或者酰胺部分相连接得到一系列新型强效的NS5A抑制剂。对核心结构各种取代模式的抑制剂进行SARs研究, 发现基于二芳基硫酸酯核心的含有酰胺部分(化合物60)或咪唑部分(化合物61)的间位双取代的抑制剂表现出极高的活性, 化合物60和61对基因1b、2a型的EC50值分别为19.8、19.9和32.3、882 pmol·L-1。并且通过hERG配体结合试验和Ames试验分别证明没有心脏毒性和诱变潜力。此外, 这些化合物与NS5B靶向药物sofosbuvir联合治疗显示出协同效应。
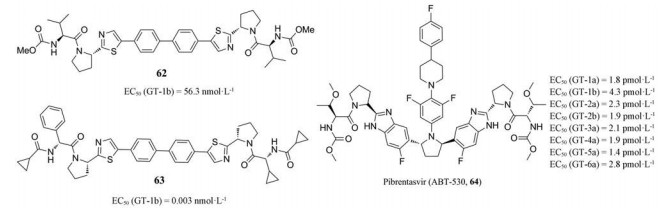
在一项初步研究中, Yeh等[71]观察到用疏水噻唑环取代DSV的两个咪唑环得到的化合物62具有中等的GT-1b抑制活性(EC50为56.3 nmol·L-1), 但是毒性小(CC50 > 50 μmol·L-1)。为了改善活性, 对其两翼进行结构修饰, 合成了一系列结构相关的二聚苯基噻唑衍生物, 并评价它们对GT-1b的抑制活性。结果表明几种化合物抑制GT-1b的活性达皮摩尔级, 其中, 环丙基酰胺衍生物63是GT-1b复制子(EC50 = 0.003 nmol·L-1)的最佳抑制剂, 并在大鼠和犬中口服给药后显示出优异PK性质。
Pibrentasvir (ABT-530, 64)是AbbVie公司继ombitasvir之后研发的第2代NS5A抑制剂, 其在ombitasvir的吡咯烷的N-1位苯基上连接了吸电性和极性都明显增强的取代基, 同时将酰胺进行成环优化形成苯并咪唑的结构。Pibrentasvir对HCV GT1-6的体外抑制活性有了进一步的提高(EC50 < 5 pmol·L-1), 同时对现有的多种NS5A抑制剂耐药变异毒株的抑制活性也在低皮摩尔水平[72]。Pibrentasvir和NS3/4A蛋白酶抑制剂glecaprevir联合应用的Ⅲ期临床试验结果表明:在1 819名患者中用药, 治疗依从性和完成度均较高(≥ 96%), 并且SVR12大于93%, 与药物相关的严重不良反应和导致治疗中断的不良反应的发生率较低(≤1%)[73]。
|
Ledipasvir、ombitasvir和daclatasvir是HCV NS5A的有效抑制剂, 其具有大分子量的杂二聚体和同二聚体结构。由于其结构复杂和难以合成, 这类HCV NS5A抑制剂具有极高成本。为了简化化学结构已设计、合成和评价了几种杂环化合物。以其中的4-苯基噻唑65 (EC50 = 9440 nmol·L-1)为先导化合物, 在65的噻唑和吡咯烷环之间引入酰胺基团得到化合物66。化合物66活性显著增加(EC50 = 0.92 nmol·L-1), 但是化合物66的PK性质一般, 并且在大鼠实验中口服生物利用度低(F = 18.7%)。进一步优化噻唑环4位和吡咯烷氮上取代基, 发现化合物67是高效和选择性NS5A抑制剂(EC50 = 4.6 nmol·L-1), 具有更高的治疗指数(SI > 10 000)。在大鼠口服给药后, 化合物67表现出良好的口服生物利用度(F = 45%)[74] (图 16)。

|
Figure 16 The structural evolution of compounds 65-67 |
PROTAC (Proteolysis targeting chimeras)分子是一种双功能小分子, 可以同时结合靶蛋白和E3-泛素连接酶, 从而引起蛋白酶对靶蛋白的泛素化和降解[75] (图 17)。与普通小分子抑制剂相比, PROTAC有以下多种优点:作用靶点广泛, 可以用于一些不可成药靶点; 用量小, 催化量的药物就可以产生很好的药理活性且选择性高; 毒副作用小, 较低的药物剂量降低了脱靶效应产生的可能性, 提高了安全性。对于抗病毒方面来说, PROTAC策略还有一个特别的优势, 通过直接将靶蛋白降解, 克服靶蛋白突变/过表达引起的耐药问题。该原理已成功应用于激酶(RIPK2、BTK、BCR-ABL和CDK9)和转录酶(BRD4、BRD9和TRIM24)等。

|
Figure 17 Schematic of the PROTAC technology |
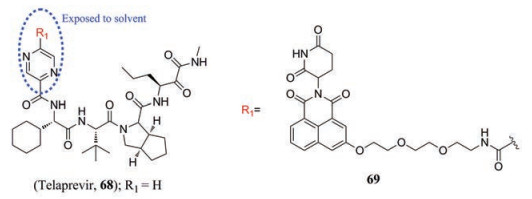
Telaprevir (VX-950, 68)是第一代NS3/4A蛋白酶抑制剂, 但是这类蛋白酶抑制剂的耐药屏障很低, 服药之后在患者体内出现多个耐药突变株。2019年, De Wispelaere等[76]选择telaprevir作为与NS3/4A蛋白酶结合的配体。通过telaprevir与蛋白酶的高分辨晶体结构发现, telaprevir的吡嗪环处于蛋白溶剂界面, 选择将吡嗪环作为与E3泛素连接酶的连接位点, 来避免连接链阻碍配体与靶蛋白之间的相互作用。基于以上研究, 他们通过使用不同的连接链来连接telaprevir与E3泛素连接酶底物CRBN, 设计了一系列小分子, 并证实了它们与NS3的靶向结合以及CRBN依懒性的降解机制。其中, 最佳化合物69能够选择性诱导快速和持续的蛋白酶体介导的NS3降解(4小时DC50为50 nmol·L-1, DC50为NS3-eGFP荧光降低50%的浓度), 此外, 化合物69不仅具有最强的抗病毒活性还保留对NS3的耐药突变株(V55A和A156S)的抑制活性。这证实了PROTAC小分子不易受配体结合位点突变的影响, 因此可以用来抑制对常规药物耐药的病毒突变株。
|
目前HCV治疗方案主要是基于几种不同作用方式的DAA组合, 组合疗法具有治愈率高、治疗周期短以及耐药屏障高等优点, 但是其价格昂贵且仍然存在耐药等问题。本文从药物化学的角度总结了治疗HCV感染的DAA研究的最新进展。基于化合物库的高通量筛选(high-throughput screening, HTS)以及从天然产物中寻找具有抗HCV活性的化合物是发现抗HCV先导化合物的重要途径。这些先导化合物基于配体结构相似性等, 通过分子杂合、生物电子等排、骨架跃迁等药物化学策略进行修饰优化; 对于已经发现的针对某确定靶点的先导化合物, 通过对其结合模式的研究, 运用基于结构的合理药物设计方法如底物包膜假说等策略进行优化; 基于先导化合物的体内代谢和活性转化的前药策略以及基于体内泛素化蛋白降解机制的PROTAC策略也用于抗HCV先导化合物的优化中。以上几种针对性修饰先导化合物的药物化学策略已经成为发展抗HCV药物的重要途径。通过系统的药代动力学、药效学和毒理学评价新衍生物, 最终筛选出候选药物。
虽然现在已经发展出许多抗HCV耐药屏障高、疗效好的临床药物, 然而, 目前针对HCV的治疗仍然存在一些挑战, 第一个挑战是对于难以治疗的人群缺少有效的治疗方案, 比如HCV感染的终末期肾病患者、肾移植前后的HCV感染患者和HCV感染的代偿性与失代偿性肝硬化患者等; 第二个是缺少耐药屏障高的泛基因型药物, 一方面是Epclusa®是唯一被批准用于治疗GT1至GT6感染的疗法; 另一方面, 耐药性仍然是一个问题, 特别是NS5A耐药相关变异对治疗结果产生了消极影响[6]。令人欣慰的是, 目前FDA批准的药物和有希望的临床前候选药物已经为大多数患者的HCV感染提供了强有力的后盾。另外, 靶向于新靶点的抑制剂, 如靶向HCV蛋白(E1/E2、p7和NS4B)与靶向人蛋白(例如亲环蛋白A、细胞周期蛋白G相关激酶和清道夫受体B1)的抑制剂; 以及具有新作用机制的抑制剂, 如PROTAC小分子, 有望解决以上难题, 这将会给HCV治疗带来新的突破。
相信在不远的将来, 随着更多高效低毒药物的出现, 全球HCV感染者将大幅减少, 根治HCV感染的目标终会全面实现。
| [1] |
World Health Organization. Hepatitis C [EB/OL]. 2018 [2019-10]. http://www.searo.who.Int/thailand/factsheets/fs0018/en/.
|
| [2] |
Alazard-Dany N, Denolly S, Boson B, et al. Overview of HCV life cycle with a special focus on current and possible future antiviral targets[J]. Viruses, 2019, 11: 30. DOI:10.3390/v11010030 |
| [3] |
Wang J, He XY. Anti-hepatitis C virus drugs: research advances[J]. J Int Pharm Res(国际药学研究杂志), 2015, 42: 551-560. |
| [4] |
Manns MP, Foster GR, Rockstroh JK, et al. The way forward in HCV treatment--finding the right path[J]. Nat Rev Drug Discov, 2007, 6: 991-1000. DOI:10.1038/nrd2411 |
| [5] |
Zając M, Muszalska I, Sobczak A, et al. Hepatitis C - new drugs and treatment prospects[J]. Eur J Med Chem, 2019, 165: 225-249. |
| [6] |
Li G, De Clercq E. Current therapy for chronic hepatitis C: The role of direct-acting antivirals[J]. Antiviral Res, 2017, 142: 83-122. |
| [7] |
Horner SM, Gale M. Regulation of hepatic innate immunity by hepatitis C virus[J]. Nat Med, 2013, 19: 879-888. DOI:10.1038/nm.3253 |
| [8] |
Hayes CN, Chayama K. Emerging treatments for chronic hepatitis C[J]. J Formos Med Assoc, 2015, 114: 204-215. DOI:10.1016/j.jfma.2014.09.001 |
| [9] |
Rusere LN, Matthew AN, Lockbaum GJ, et al. Quinoxaline-based linear HCV NS3/4A protease inhibitors exhibit potent activity against drug resistant variants[J]. ACS Med Chem Lett, 2018, 9: 691-696. DOI:10.1021/acsmedchemlett.8b00150 |
| [10] |
Wang AX, Chen J, Zhao Q, et al. Structure-activity relationships of 4-hydroxy-4-biaryl-proline acylsulfonamide tripeptides: a series of potent NS3 protease inhibitors for the treatment of hepatitis C virus[J]. Bioorg Med Chem Lett, 2017, 27: 590-596. DOI:10.1016/j.bmcl.2016.12.013 |
| [11] |
Scola PM, Sun LQ, Wang AX, et al. The discovery of asunaprevir (BMS-650032), an orally efficacious NS3 protease inhibitor for the treatment of hepatitis C virus infection[J]. J Med Chem, 2014, 57: 1730-1752. DOI:10.1021/jm500297k |
| [12] |
Gising J, Belfrage AK, Alogheli H, et al. Achiral pyrazinone-based inhibitors of the hepatitis C virus NS3 protease and drug-resistant variants with elongated substituents directed toward the S2 pockect[J]. J Med Chem, 2014, 57: 1790-1801. DOI:10.1021/jm301887f |
| [13] |
Belfrage AK, Abdurakhmanov E, Åkerblom E, et al. Pan-NS3 protease inhibitors of hepatitis C virus based on an R3-elongated pyrazinone scaffold[J]. Eur J Med Chem, 2018, 148: 453-464. DOI:10.1016/j.ejmech.2018.02.032 |
| [14] |
Zheng B, D'Andrea SV, Sun LQ, et al. Potent inhibitors of hepatitis C virus NS3 protease: employment of a difluoromethyl group as a hydrogen-bond donor[J]. ACS Med Chem Lett, 2018, 9: 143-148. DOI:10.1021/acsmedchemlett.7b00503 |
| [15] |
Shah U, Jayne C, Chackalamannil S, et al. Novel quinoline-based P2-P4 macrocyclic derivatives as pan-genotypic HCV NS3/4a protease inhibitors[J]. ACS Med Chem Lett, 2014, 5: 264-269. DOI:10.1021/ml400466p |
| [16] |
Neelamkavil SF, Agrawal S, Bara T, et al. Discovery of MK-8831, a novel spiro-proline macrocycle as a pan-genotypic HCV-NS3/4a protease inhibitor[J]. ACS Med Chem Lett, 2015, 7: 111-116. |
| [17] |
Velázquez F, Chelliah M, Clasby M, et al. Design and synthesis of P2-P4 macrocycles containing a unique spirocyclic proline: a new class of HCV NS3/4A inhibitors[J]. ACS Med Chem Lett, 2016, 7: 1173-1178. DOI:10.1021/acsmedchemlett.6b00321 |
| [18] |
Pawlotsky JM. Hepatitis C virus resistance to direct-acting antiviral drugs in interferon-free regimens[J]. Gastroenterology, 2016, 151: 70-86. DOI:10.1053/j.gastro.2016.04.003 |
| [19] |
Matthew AN, Zephyr J, Hill CJ, et al. Hepatitis C virus NS3/4A protease inhibitors incorporating flexible P2 quinoxalines target drug resistant viral variantsc[J]. J Med Chem, 2017, 60: 5699-5716. DOI:10.1021/acs.jmedchem.7b00426 |
| [20] |
Yang H, Yang C, Wang Y, et al. Preclinical characterization of the novel HCV NS3 protease inhibitor GS-9256[J]. Antivir Ther, 2017, 22: 413-420. DOI:10.3851/IMP3132 |
| [21] |
Sheng XC, Casarez A, Cai RC, et al. Discovery of GS-9256: a novel phosphinic acid derived inhibitor of the hepatitis C virus NS3/4A protease with potent clinical activity[J]. Bioorg Med Chem Lett, 2012, 22: 1394-1396. |
| [22] |
Bowsher M, Hiebert S, Li R, et al. The discovery and optimization of naphthalene-linked P2-P4 Macrocycles as inhibitors of HCV NS3 protease[J]. Bioorg Med Chem Lett, 2018, 28: 43-48. DOI:10.1016/j.bmcl.2017.11.005 |
| [23] |
Watkins WJ, Ray AS, Chong LS. HCV NS5B polymerase inhibitors[J]. Curr Opin Drug Discov Devel, 2010, 13: 441-465. |
| [24] |
Membreno FE, Lawitz EJ. The HCV NS5B nucleoside and non-nucleoside inhibitors[J]. Clin Liver Dis, 2011, 15: 611-626. DOI:10.1016/j.cld.2011.05.003 |
| [25] |
Hedskog C, Dvory-Sobol H, Gontcharova V, et al. Evolution of the HCV viral population from a patient with S282T detected at relapse after sofosbuvir monotherapy[J]. J Viral Hepat, 2015, 22: 871-881. |
| [26] |
Zhen L, Dai L, Wen X, et al. Discovery of novel nucleotide prodrugs with improved potency against HCV variants carrying NS5B S282T mutation[J]. J Med Chem, 2017, 60: 6077-6088. |
| [27] |
Zhou S, Mahmoud S, Liu P, et al. 2'-Chloro, 2'-fluoro ribonucleotide prodrugs with potent pan-genotypic activity against hepatitis C virus replication in culture[J]. J Med Chem, 2017, 60: 5424-5437. |
| [28] |
Jonckers TH, Vandyck K, Vandekerckhove L, et al. Nucleotide prodrugs of 2'-deoxy-2'-spirooxetane ribonucleosides as novel inhibitors of the HCV NS5B polymerase[J]. J Med Chem, 2014, 57: 1836-1844. |
| [29] |
Jonckers TH, Tahri A, Vijgen L, et al. Discovery of 1-((2R, 4aR, 6R, 7R, 7aR)-2-isopropoxy-2-oxidodihydro-4H, 6H-spiro[furo[3, 2-d][1, 3, 2]dioxaphosphinine-7, 2'-oxetan]-6-yl)pyrimidine-2, 4(1H, 3H)-dione (JNJ-54257099), a 3'-5'-cyclic phosphate ester prodrug of 2'-deoxy-2'-spirooxetane uridine triphosphate useful for HCV inhibition[J]. J Med Chem, 2016, 59: 5790-5798. DOI:10.1021/acs.jmedchem.6b00382 |
| [30] |
Cheng Y, Shen J, Peng RZ, et al. Structure-based optimization and derivatization of 2-substituted quinolone-based non-nucleoside HCV NS5B inhibitors with submicromolar cellular replicon potency[J]. Bioorg Med Chem Lett, 2016, 26: 2900-2906. DOI:10.1016/j.bmcl.2016.04.042 |
| [31] |
Manfroni G, Cannalire R, Barreca ML, et al. The versatile nature of the 6-aminoquinolone scaffold: identification of submicromolar hepatitis C virus NS5B inhibitors[J]. J Med Chem, 2014, 57: 1952-1963. |
| [32] |
Court JJ, Poisson C, Ardzinski A, et al. Discovery of novel thiophene-based, thumb pocket 2 allosteric inhibitors of the hepatitis C NS5B polymerase with improved potency and physicochemical profiles[J]. J Med Chem, 2016, 59: 6293-6302. |
| [33] |
Li P, Dorsch W, Lauffer DJ, et al. Discovery of novel allosteric HCV NS5B inhibitors. 2. Lactam-containing thiophene carboxylates[J]. ACS Med Chem Lett, 2017, 8: 251-255. DOI:10.1021/acsmedchemlett.6b00479 |
| [34] |
Barnes-Seeman D, Boiselle C. Design and synthesis of lactam-thiophene carboxylic acids as potent hepatitis C virus polymerase inhibitors[J]. Bioorg Med Chem Lett, 2014, 24: 3979-3985. DOI:10.1016/j.bmcl.2014.06.031 |
| [35] |
Yeung KS, Beno BR, Parcella K, et al. Discovery of a hepatitis C Virus NS5B replicase palm site allosteric inhibitor (BMS-929075) advanced to Phase 1 clinical studies[J]. J Med Chem, 2017, 60: 4369-4385. |
| [36] |
Eastman KJ, Parcella K, Yeung KS, et al. The discovery of a pan-genotypic, primer grip inhibitor of HCV NS5B polymerase[J]. Med Chem Commun, 2017, 8: 796-806. DOI:10.1039/C6MD00636A |
| [37] |
Parcella K, Eastman K, Yeung KS, et al. Improving metabolic stability with deuterium: the discovery of BMT-052, a pan-genotypic HCV NS5B polymerase inhibitor[J]. ACS Med Chem Lett, 2017, 8: 771-774. DOI:10.1021/acsmedchemlett.7b00211 |
| [38] |
Meguellati A, Ahmed-Belkacem A, Nurisso A, et al. New pseudodimeric aurones as palm pocket inhibitors of hepatitis C virus RNA-dependent RNA polymerase[J]. Eur J Med Chem, 2016, 115: 217-229. DOI:10.1016/j.ejmech.2016.03.005 |
| [39] |
Randolph JT, Krueger AC, Donner PL, et al. Synthesis and biological characterization of aryl uracil inhibitors of hepatitis C virus NS5B polymerase: discovery of ABT-072, a trans-stilbene analog with good oral bioavailability[J]. J Med Chem, 2018, 61: 1153-1163. DOI:10.1021/acs.jmedchem.7b01630 |
| [40] |
Camarasa M, Puig de la Bellacasa R, González ÀL, et al. Design, synthesis and biological evaluation of pyrido[2, 3-d]pyrimidin-7-(8H)-ones as HCV inhibitors[J]. Eur J Med Chem, 2016, 115: 463-483. DOI:10.1016/j.ejmech.2016.03.055 |
| [41] |
Kaushik-Basu N, Ratmanova NK, Manvar D, et al. Bicyclic octahydrocyclohepta [b]pyrrol-4(1H)-one derivatives as novel selective anti-hepatitis C virus agents[J]. Eur J Med Chem, 2016, 122: 319-325. DOI:10.1016/j.ejmech.2016.06.041 |
| [42] |
Belema M, Meanwell NA. Discovery of daclatasvir, a pan-genotypic hepatitis C virus NS5A replication complex inhibitor with potent clinical effect[J]. J Med Chem, 2014, 57: 5057-5071. DOI:10.1021/jm500335h |
| [43] |
Bachand C, Belema M, Deon DH, et al. Hepatitis C virus inhibitors: US, 021927 [P]. 2008-11-27.
|
| [44] |
Bachand C, Belema M, Deon DH, et al. Hepatitis C virus inhibitors: US, 021928 [P]. 2008-12-04.
|
| [45] |
Bachand C, Belema M, Deon DH, et al. Hepatitis C virus inhibitors: US, 021936 [P]. 2008-12-18.
|
| [46] |
Li L, Zhong M. Inhibitors of HCV NS 5A: US, 065668 [P]. 2010-06-26.
|
| [47] |
Zhong M, Li L. Inhibitors of HCV NS 5A: US, 102010 [P]. 2010-06-10.
|
| [48] |
Li L, Zhong M. Inhibitors of HCV NS 5A: US, 065681 [P]. 2008-12-03.
|
| [49] |
Guo H, Kato D, Kirschberg TA, et al. Antiviral compounds: US, 092010 [P]. 2010-12-09.
|
| [50] |
Belema M, Lopez OD, Bender JA, et al. Discovery and development of hepatitis C virus NS5A replication complex inhibitors[J]. J Med Chem, 2014, 57: 1643-1672. |
| [51] |
Ivachtchenko AV, Mitkin OD, Yamanushkin PM, et al. Discovery of novel highly potent hepatitis C virus NS5A inhibitor (AV4025)[J]. J Med Chem, 2014, 57: 7716-7730. DOI:10.1021/jm500951r |
| [52] |
DeGoey DA, Randolph JT, Liu D, et al. Discovery of ABT-267, a pan-genotypic inhibitor of HCV NS5A[J]. J Med Chem, 2014, 57: 2047-2057. DOI:10.1021/jm401398x |
| [53] |
Franz AK, Wilson SO. Organosilicon molecules with medicinal applications[J]. J Med Chem, 2012, 56: 388-405. |
| [54] |
Lazareva N, Lazarev I. Drug design based on the carbon/silicon switch strategy[J]. Russ Chem Bull, 2015, 64: 1221-1232. DOI:10.1007/s11172-015-1005-4 |
| [55] |
Nair AG, Zeng Q, Selyutin O, et al. Discovery of silyl proline containing HCV NS5A inhibitors with pan-genotype activity: SAR development[J]. Bioorg Med Chem Lett, 2016, 26: 1475-1479. |
| [56] |
Liu B, Gai K, Qin H, et al. Design, synthesis and identification of silicon-containing HCV NS5A inhibitors with pan-genotype activity[J]. Eur J Med Chem, 2018, 148: 95-105. |
| [57] |
Coburn CA, Meinke PT, Chang W, et al. Discovery of MK-8742: an HCV NS5A inhibitor with broad genotype activity[J]. ChemMedChem, 2013, 8: 1930-1940. DOI:10.1002/cmdc.201300343 |
| [58] |
Rockstroh JK, Nelson M, Katlama C, et al. Efficacy and safety of grazoprevir (MK-5172) and elbasvir (MK-8742) in patients with hepatitis C virus and HIV co-infection (C-EDGE CO-INFECTION): a non-randomised, open-label trial[J]. Lancet HIV, 2015, 2: e319-27. |
| [59] |
Tong L, Yu W, Chen L, et al. Discovery of ruzasvir (MK-8408): a potent, pan-genotype HCV NS5A inhibitor with optimized activity against common resistance-associated polymorphisms[J]. J Med Chem, 2017, 60: 290-306. DOI:10.1021/acs.jmedchem.6b01310 |
| [60] |
Lawitz E, Gane E, Feld JJ, et al. Efficacy and safety of a two-drug direct-acting antiviral agent regimen ruzasvir 180 mg and uprifosbuvir 450 mg for 12 weeks in adults with chronic hepatitis C virus genotype 1, 2, 3, 4, 5 or 6[J]. J Viral Hepat, 2019, 26: 1127-1138. |
| [61] |
Kazmierski WM, Maynard A, Duan M, et al. Novel spiroketal pyrrolidine GSK2336805 potently inhibits key hepatitis C virus genotype 1b mutants: from lead to clinical compound[J]. J Med Chem, 2014, 57: 2058-2073. DOI:10.1021/jm4013104 |
| [62] |
Vijgen L, Thys K, Vandebosch A, et al. Virology analysis in HCV genotype 1-infected patients treated with the combination of simeprevir and TMC647055/ritonavir, with and without ribavirin, and JNJ-56914845[J]. Virol J, 2017, 14: 101. DOI:10.1186/s12985-017-0760-2 |
| [63] |
Dwyer MP, Keertikar KM, Chen L, et al. Matched and mixed cap derivatives of the HCV NS5A inhibitor MK-8742[J]. Bioorg Med Chem Lett, 2016, 26: 4106-4111. DOI:10.1016/j.bmcl.2016.06.063 |
| [64] |
Yu W, Zhou G, Coburn CA, et al. Substituted tetracyclic indole core derivatives of HCV NS5A inhibitor MK-8742[J]. Bioorg Med Chem Lett, 2016, 26: 4851-4856. DOI:10.1016/j.bmcl.2016.08.002 |
| [65] |
Tong L, Yu W, Coburn CA, et al. Structure-activity relationships of proline modifications around the tetracyclic-indole class of NS5A inhibitors[J]. Bioorg Med Chem Lett, 2016, 26: 5354-5360. |
| [66] |
Yu W, Tong L, Hu B, et al. Discovery of chromane containing HCV NS5A inhibitors with improved potency against resistance associate variants[J]. J Med Chem, 2016, 59: 10228-10243. DOI:10.1021/acs.jmedchem.6b01234 |
| [67] |
Yu W, Tong L, Selyutin O, et al. Discovery of MK-6169, a potent pan-genotype hepatitis C virus NS5A inhibitor with optimized activity against common resistance-associated substitutions[J]. J Med Chem, 2018, 61: 3984-4003. DOI:10.1021/acs.jmedchem.7b01927 |
| [68] |
Bae IH, Choi JK, Chough C, et al. Potent hepatitis C virus NS5A inhibitors containing a benzidine core[J]. ACS Med Chem Lett, 2013, 5: 255-258. |
| [69] |
Bae IH, Kim HS, You Y, et al. Novel benzidine and diaminofluorene prolinamide derivatives as potent hepatitis C virus NS5A inhibitors[J]. Eur J Med Chem, 2015, 101: 163-178. DOI:10.1016/j.ejmech.2015.06.033 |
| [70] |
You Y, Kim HS, Bae IH, et al. New potent biaryl sulfate-based hepatitis C virus inhibitors[J]. Eur J Med Chem, 2017, 125: 87-100. DOI:10.1016/j.ejmech.2016.09.031 |
| [71] |
Yeh TK, Kang IJ, Hsu TA, et al. A novel, potent, and orally bioavailable thiazole HCV NS5A inhibitor for the treatment of hepatitis C virus[J]. Eur J Med Chem, 2019, 167: 245-268. |
| [72] |
Wagner R, Randolph JT, Patel SV, et al. Highlights of the structure-activity relationships of benzimidazole linked pyrrolidines leading to the discovery of the hepatitis C virus NS5A inhibitor pibrentasvir (ABT-530)[J]. J Med Chem, 2018, 61: 4052-4066. DOI:10.1021/acs.jmedchem.8b00082 |
| [73] |
Foster GR, Dore GJ, Wang S, et al. Glecaprevir/pibrentasvir in patients with chronic HCV and recent drug use: an integrated analysis of 7 phase Ⅲ studies[J]. Drug Alcohol Depend, 2019, 194: 487-494. DOI:10.1016/j.drugalcdep.2018.11.007 |
| [74] |
Kang IJ, Hsu SJ, Yang HY, et al. Potent, selective, and orally bioavailable HCV NS5A inhibitor for treatment of hepatitis C virus: (S)-1-((R)-2-(cyclopropanecarboxamido)-2-phenylacetyl)-N-(4-phenylthiazol-2-yl)pyrrolidine-2-carboxamide[J]. J Med Chem, 2017, 60: 228-247. DOI:10.1021/acs.jmedchem.6b00962 |
| [75] |
Neklesa TK, Winkler JD, Crews CM. Targeted protein degradation by PROTACs[J]. Pharmacol Ther, 2017, 174: 138-144. |
| [76] |
De Wispelaere M, Du G, Donovan KA, et al. Small molecule degraders of the hepatitis C virus protease reduce susceptibility to resistance mutations[J]. Nat Commun, 2019, 10: 3468. DOI:10.1038/s41467-019-11429-w |