 2020, Vol. 55
2020, Vol. 55



寨卡病毒(Zika virus, ZIKV)是黄病毒科黄病毒属的成员之一[1], 最早于1947年分离自乌干达寨卡森林一只恒河猴的血清样本[2]。1948年, 从同一森林的非洲伊蚊中也分离出这种病毒。在20世纪50年代, 首次报告了人类感染ZIKV的病例[3]。2015年5月, ZIKV横贯太平洋, 在巴西引起100多万感染病例[4]。截至2019年5月, 该病毒迅速传播到84个国家和地区, 已经成为世界范围内的公共卫生问题[5]。
ZIKV的遗传物质为单股正链RNA, 复制周期类似于其他已知的黄病毒(图 1)。首先, 病毒附着到宿主细胞膜上的受体, 在低pH环境下, 通过网格蛋白介导内吞[6]。在此过程中, 酪氨酸蛋白激酶受体AXL、Tyro 3、DC-SIGN和Tim-1等细胞表面受体促进了ZIKV进入宿主细胞[7]。接着, 病毒基因组被释放到宿主的细胞质中进行翻译, 由此产生的多聚蛋白水解为各种蛋白质, 包括结构和非结构蛋白[8, 9]。同时, 新合成的正链RNA可以用于进一步的翻译或复制, 也可以在内质网中进行组装, 并通过分泌通路, 再成为成熟的子代病毒, 最后释放到细胞外间隙。

|
Figure 1 The life cycle of Zika virus (ZIKV) |
ZIKV的遗传物质单股正链RNA长度为10.8 kb, 由约100个核苷酸长度的5′端非翻译区, 10 kb的开放阅读框和约420个核苷酸长度的3′端非翻译区组成。开放阅读框编码了长度为3 423个氨基酸的多聚蛋白质前体。多聚蛋白质前体经共翻译转运及后翻译转运, 水解为3种结构蛋白(C、prM和E)以及8种非结构蛋白(NS1、NS2A、NS2B、NS3、S4A、2K、NS4B和NS5)[10] (图 2)。

|
Figure 2 Detailed structure of ZIKV genome |
三种结构蛋白负责形成病毒粒子, 并参与病毒进入、组装和释放到宿主细胞中。衣壳蛋白结合基因组RNA形成核衣壳, E和prM糖蛋白是连接宿主细胞膜的病毒表面蛋白。
非结构蛋白在宿主细胞内形成病毒复制复合体[10]。NS3和NS5蛋白具有酶活性。NS3编码丝氨酸蛋白酶[11, 12]、RNA解旋酶[13]、核苷三磷酸(NTPase)[14]和RNA三磷酸酶(RTPase)[15]酶活性。在黄病毒中, NS3具有良好的保守性, 寨卡病毒(ZIKV)、西尼罗河病毒(WNV)、日本脑炎病毒(JEV)和黄热病病毒(YFV)之间的氨基酸序列同源性约为65%。NS3蛋白含有两个结构域, 即N端蛋白酶结构域和C端螺旋酶结构域, 二者通过短链连接。由于NS3蛋白在病毒生命周期中扮演了多重角色, 该蛋白是抗病毒药物发现的热门靶点。NS5编码甲基转移酶(MTase)和鸟苷酰转移酶(GTase)以及RNA依赖的RNA聚合酶(RdRp)[16]。NS2A、NS2B、NS4A、2K和NS4B是位于内质网的跨膜蛋白[17]。NS2B、NS4A和NS4B的一些区域也与NS3相互作用, 从而锚定由NS3和NS5形成的复制复合物到内质网[18]。此外, NS2B也是NS3蛋白酶和NS4B阻断干扰素α/β信号转导的重要辅助因子[19]。NS4A诱导内质网重排, 从而参与形成病毒复制区[20]。NS1通过调节宿主防御机制以达到免疫逃避作用, 因此同样被认为参与了基因组复制[21]。
到目前为止, 还没有特定的抗病毒药物被批准用于治疗ZIKV感染。临床上多使用常用药物来缓解症状, 比如对乙酰氨基酚通常用于控制发烧和疼痛, 抗组胺药物用于瘙痒皮疹, 而阿司匹林等非甾体抗炎药物(NSAIDs)由于出血综合征和瑞夷综合征风险增加而被禁止。
目前ZIKV抑制剂的发现策略主要包括:筛选多样性的小分子化合物库或天然化合物[22, 23], 以及已知活性化合物或临床应用药物进行“药物重定位”。此外, 基于抗体的候选药物研究[24, 25]也是一个重要方面。在过去的几年里, 大量的先导化合物和候选药物被开发出来, 其中有的已进入临床试验。按照其作用方式可分为作用于病毒本身的药物及作用于宿主细胞的药物。
1 作用于病毒的药物此类药物直接针对病毒靶标, 可分为: NS5 RdRp抑制剂; NS5 MTase抑制剂; NS2B-NS3蛋白酶抑制剂和NS3螺旋酶抑制剂。
1.1 NS5 RdRp抑制剂ZIKV的RdRp为右旋构象, 由3部分构成(图 3):手指部分(α1~α9, α12~β3)、手掌部分(α10、α11、310螺旋结构η1、α14~310螺旋结构η2)以及拇指部分(β6~α23)[26]。

|
Figure 3 Structure of ZIKV NS5 RdRp (Contains two zinc-binding pockets) |
2016年, Eyer等[27]测试了29种核苷类似物(浓度为50 μmol·L-1)在Vero细胞中抑制细胞病变效应(CPE)的能力, 发现其中5种化合物即7-二氮-2′-C-甲基腺苷(7-deaza-2′-CMA)、2′-C-甲基腺苷(2-CMA)、2′-C-甲基胞苷(2-CMC)、2′-C-甲基鸟嘌呤(2-CMG)和2′-C-甲基尿苷(2-CMU)可明显降低ZIKV感染细胞的死亡率, 其半数有效浓度(EC50)为5.3~45.5 μmol·L-1。在检测不同的2′-C甲基化核苷对纯化后的重组ZIKV RdRp的体外活性的影响时, 也得到了类似的结果。此外, 病毒聚合酶抑制剂7-deaza-2′-CMA在Vero细胞中也具有抗ZIKV活性(EC50 = 9.6 μmol·L-1), 在50 mg·kg-1·d-1剂量下, 可延缓ZIKV感染的AG 129 (干扰素-α/β和IFN-γ受体基因敲除)小鼠的发病率和死亡率, 降低病毒血症发生率。尽管7-deaza-2′-CMA抑制剂在治疗慢性丙型肝炎的临床试验中失败(可能由于线粒体毒性[28]), 该化合物仍然适用于短期治疗急性黄病毒疾病, 包括WNV[29], 也是迄今为止治疗黄病毒感染最有希望的候选药物之一。
法匹拉韦(6, favipiravir, 6-氟-3-羟基-2-吡嗪甲酰胺), 原名T-705, 是一种新型的抗病毒药物, 可选择性地有效抑制黄病毒、正粘病毒、甲病毒、线状病毒、布尼亚病毒、沙粒病毒、杯状病毒、诺如病毒和其他RNA病毒的RdRp[30]。法匹拉韦与RdRp作用的确切分子机制尚未完全阐明, 目前的假说主要是法匹拉韦可能掺入新生的病毒RNA中, 或者通过与保守的聚合酶结构域结合, 从而阻止核苷酸在病毒RNA复制和转录中与模板结合以达到治疗效果。2017年, 法匹拉韦对多个ZIKV毒株的抑制作用在体外实验中已经被Baz和Cai等证明(EC50 = 35 μmol·L-1)[31, 32]。此外, 法匹拉韦对于多种病毒的耐药基因屏障也较高。在健康志愿者和流感患者参与临床试验中, 法匹拉韦展示出良好的耐受性。然而, 由于这种药物存在致畸风险, 仍需谨慎使用[33]。
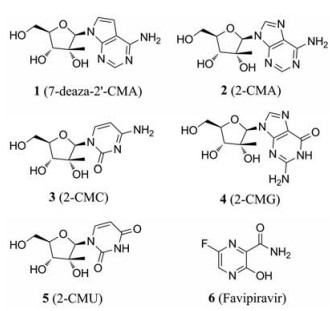
|
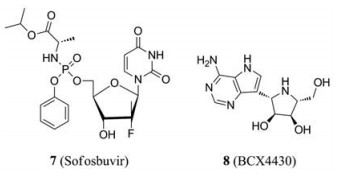
索非布韦(7, sofosbuvir)是一种核苷类似物, 可用于治疗慢性HCV感染, 属于RdRp抑制剂。索非布韦氨基磷酸酯作为前药, 在细胞环境中转化为三磷酸类似物从而产生活性。其活性代谢物2′-氟-2-C-甲基-UTP与NS5的活性位点结合[34], 具有抑制ZIKV感染和复制的作用。索非布韦在人肝癌细胞(Huh-7)、人胎盘绒毛膜癌细胞(Jar)、SH-Sy5y神经母细胞瘤细胞中的EC50值在0.4~5 μmol·L-1范围内, 在人胚胎后脑和大脑皮层神经元干细胞(NSCs)中的EC50约32 μmol·L-1 [34-36], 但在Vero细胞中未表现出抗ZIKV活性。因此, 索非布韦在不同细胞类型中抑制ZIKV活性不同[36]。另外, 从使用索非布韦的受感染细胞中分离出的ZIKV序列分析表明, 与未处理组相比, ZIKV的突变频率更高[36], 这表明, 除了其抑制作用外, 该药物还能增加病毒基因组中突变的发生, 增加复制的出错率[37]。索非布韦以33 mg·kg-1·d-1的剂量, 口服给与抗干扰素α受体1 (IFN-αR1)封闭抗体治疗的野生型C57BL/6小鼠7天[38], 可保护50%的小鼠免受ZIKV感染引起的体重减轻和死亡。然而, 较高浓度的药物无效, 还会产生毒性[35]。值得关注的是, 在Ⅱ期和Ⅲ期临床研究中, 索非布韦在治疗HCV感染方面被证明是安全和有效的[39]。
BCX 4430属于腺苷类似物, 是病毒RdRp的选择性抑制剂。该化合物对RNA病毒具有广谱活性, 包括WNV、YFV、马尔堡病毒和埃博拉病毒[40, 41]。BCX 4430作用于NS5聚合酶, 促进病毒RNA合成的链终止[38], 在Vero细胞中抑制ZIKV复制的EC50为3.8~11.7 μg·mL-1, 根据病毒株的不同, 选择指数分别为5.5和11.6。此外, BCX 4430以300 mg·kg-1的剂量肌肉注射, 每日两次, 可治疗AG 129小鼠ZIKV感染(马来西亚株, P-6-740), 治疗后24 h可显著降低病毒血症, 避免87.5%的小鼠死亡[42]。该化合物目前正处于Ⅰ期临床试验阶段。
1.2 NS5 MTase抑制剂ZIKV的MTase为同型二聚体, 由两个构象不同的相同亚基构成(图 4A)[43]。
|

|
Figure 4 Structure and potential ligand binding sites of ZIKV NS5 MTase |
2016年, Zhang等[44]使用FTmap program和metaPocket 2.0 server, 预测了MTase的3个潜在靶点(图 4B)。GTP/RNA帽结合位点为Site 1和Site 2, Site 3具有疏水性和极性特点, 适合通过高通量筛选发现小分子化合物。
西奈芬净(9, sinefungin)是一种腺苷衍生物, 属于泛甲基转移酶抑制剂, 最初作为一种潜在的抗真菌药物从灰质链霉菌中分离出来[45]。Hercik等[46]报道了西奈芬净与ZIKV甲基转移酶所形成配合物的晶体结构。SAM为MTase的天然底物, 西奈芬净是其竞争性抑制剂[47]。西奈芬净与GTP和GDP类似物相结合, 可能增强这些类似物对酶的亲和力, 从而得到更好的选择性和对ZIKV复制更强的抑制作用[46]。然而, 西奈芬净作为抗寄生虫剂在犬和山羊上进行实验时, 被发现具有毒性, 阻碍了其临床应用[48]。
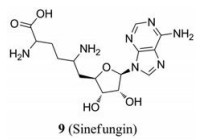
|
NS2B-NS3胰蛋白酶样丝氨酸蛋白酶在病毒复制过程中起着关键作用。2016年, Zhang等[49]报道了自由状态下ZIKV非连接NS2B:NS3蛋白酶(bZiPro)及其底物结合口袋的结构(图 5)。

|
Figure 5 The free-enzyme form of bZiPro and substrate binding pocket |
2016年, Nitsche等[50]合成了8种肽-硼酸类抑制剂, 利用bZiPro测试后, 发现其中化合物10 (EC50 = 0.25 μmol·L-1)和11 (EC50 = 0.83 μmol·L-1)活性较好。同年, Lei等[51]进一步研究了化合物10与bZiPro的结合情况(图 6)。

|
Figure 6 The structures of compounds 10 and 11, and the binding mode of compound 10 with bZiPro |
2017年, Lee等[52]通过对约40 000个化合物进行高通量筛选, 初步发现了10个磺酰胺类化合物(12~21)对ZIKV的NS2B-NS3蛋白酶具有抑制活性(EC50 < 50 μmol·L-1)和结合活性(KD约5~10 μmol·L-1)。
2017年, Li等[53]通过使用bZiPro测试, 报道了一种活性较高(EC50 = 208 nmol·L-1)的二肽类抑制剂22, 该抑制剂与NS2B-NS3蛋白酶135位丝氨酸共价连接, 其赖氨酸和精氨酸残基则分别结合于酶的S1和S2口袋; 2018年, 该课题组又应用同样的研究方法报道了一种小分子变构抑制剂23[54] (EC50 = 1.5 μmol·L-1), 该抑制剂通过与51位组氨酸形成氢键及与161位酪氨酸形成π-π叠加作用, 从而稳定了酶的闭合构象(图 7)。
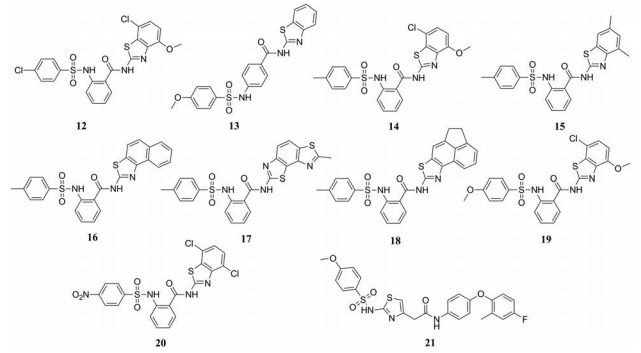
|

许多天然化合物, 如多酚, 具有NS2B-NS3蛋白酶抑制作用及抗ZIKV活性。2017年, Roy等[55]测试了杨梅酮(myricetin)、栎皮酮(quercetin)、木犀草素(luteolin)、异鼠李素(isorhamnetin)、芹菜素(apigenin)和姜黄素(curcumin) 6种天然化合物, 发现它们具有ZIKV的NS2B-NS3蛋白酶抑制活性。该类化合物可与活性位点背面的口袋结合, 通过变构效应影响NS2B-NS3蛋白酶的催化活性。前5种黄酮类化合物的EC50范围为1.3~56.3 μmol·L-1, 姜黄素EC50为3.5 μmol·L-1。
2017年, Li等[56]通过筛选2 816种FDA已上市或在研药物, 发现23种化合物抑制ZIKV的EC50低于15 μmol·L-1。然而, 这些化合物中有12种被认为是假阳性化合物(PAINS)。其余化合物中有3种的EC50值在1.1到15.9 μmol·L-1范围内, 分别为替莫泊芬(30, Temoporfin)、氯硝柳胺(31, Niclosamide)和硝唑尼特(32, Nitazoxanide)。替莫泊芬显示极低的EC50值(纳摩尔范围), 在致死小鼠模型中测试时, 能够抑制病毒血症并可保护83%的受感染小鼠。此外, 幸存下来的小鼠并没有出现任何神经紊乱的迹象。氯硝柳胺是FDA批准的肠道蠕虫病治疗药物, 已经使用了50多年, 安全性好。这种药物可阻止内体的酸化, 但其具体机制尚未阐明[57]。根据细胞内病毒RNA的测定, 氯硝柳胺在胶质母细胞瘤SNB-19细胞中抑制ZIKV的EC50为0.37 μmol·L-1 [58]。
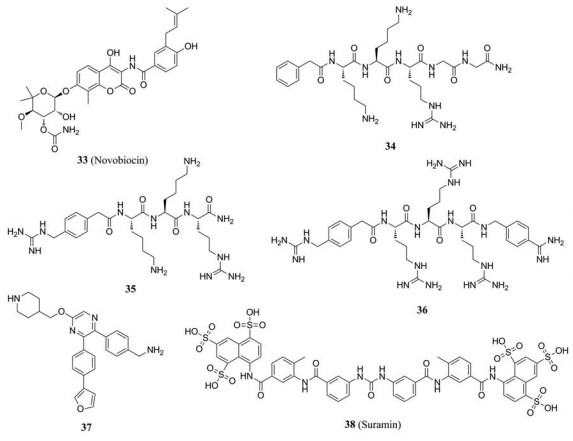
同年, Yuan等[59]通过大型化学库(8 277种化合物)的虚拟筛选, 成功发现了8种具有抑制ZIKV NS2B-NS3蛋白酶活性的化合物。此外, 还通过体外抗病毒试验和动物实验进一步验证了新生霉素(33, Novobiocin)的抗ZIKV活性。在体外抗病毒试验中, 新生霉素的EC50值为24.82 μmol·L-1。在动物实验中, 感染后第1天至第13天, 以100 mg·kg-1 (一天两次)的剂量给予免疫缺陷小鼠以新生霉素, 测得EC50值为24.82 μmol·L-1。此外, 小鼠存活率由0%提高至100%, 平均血和组织病毒载量下降, 且与未经治疗的对照组相比, 组织病理学改变较轻。

|
2018年, Phoo等[60]通过使用bZiPro测试, 报道了3种活性较高的拟肽类抑制剂(34~36), EC50分别为1.2、1.6和1.1 μmol·L-1, Ki分别为0.047、0.06和0.43 μmol·L-1。
2019年, Yuan等[61]利用重组ZIKV蛋白酶, 对1 200种化合物进行筛选及结构优化, 得到了活性较高的化合物37 (EC50 = 0.2 μmol·L-1)。细胞实验显示, 37的活性呈剂量相关, 剂量为300 nmol·L-1时, 可使被感染的U87神经胶质瘤细胞中ZIKV数量下降68%;剂量为2.5 μmol·L-1时, ZIKV数量可下降99%。动物实验显示, 以15 mg·kg-1·112 h-1的剂量给予ZIKV感染的C57BL/6小鼠, 24 h后, 其血浆及大脑中ZIKV量分别减少96%及98%。
1.4 NS3螺旋酶抑制剂NS3螺旋酶具有腺苷三磷酸酶(ATPase)和RNA三磷酸酶(RTPase)活性。NS3抑制剂可抑制ZIKV感染。2016年, Jain等[62]报道了NS3螺旋酶的结构(图 8), 发现黄病毒的NS3螺旋酶结构均具有相似性, 而差异性则主要表现在静电表面的差别、Domain 1与2的相对方位及ATP和RNA结合部位的构造方面。

|
Figure 8 Structure of ZIKV NS3 helicase[63] |
苏拉明(38, Suramin), 一种用于治疗抗寄生虫药物。该药物还能抑制多种DNA和RNA病毒, 包括DENV、1型单纯疱疹病毒、巨细胞病毒、肝炎病毒(乙型、丙型、丁型)、布尼亚病毒等[63-66]。Albulescu等[67]研究发现, 苏拉明可干扰病毒附着和子代ZIKV释放, 从而具有抗ZIKV活性, EC50为39.8 μmol·L-1。当病毒进入宿主细胞后, 其RNA合成不受苏拉明影响, 但ZIKV基因组的释放速率以及传染性均降低, 提示该药物可能通过干扰ZIKV在分泌过程中的糖基化和成熟来发挥作用。
|
2019年, Zhu等[68]通过对1974种化合物进行高通量筛选, 发现NSC10580和NSC45741两种化合物(39、40)对NS3螺旋酶具有较好抑制活性, 在BHK-21细胞系上抗ZIKV感染的EC50分别为8.5和15.2 μmol·L-1, 故其可成为抗ZIKV先导化合物。
2 作用于宿主细胞的药物此类药物作用于宿主细胞, 可分为:嘧啶合成抑制剂、嘌呤合成抑制剂、次黄嘌呤核苷酸脱氢酶(IMPDH)抑制剂、3-羟基-3-甲基戊二酸单酰辅酶A (HMG-CoA)还原酶抑制剂、针对囊泡ATP酶的病毒侵入阻断剂、病毒复制pH依赖步骤抑制剂、细胞周期依赖性激酶抑制剂、神经细胞死亡抑制剂和Bcl-2蛋白抑制剂等类型。
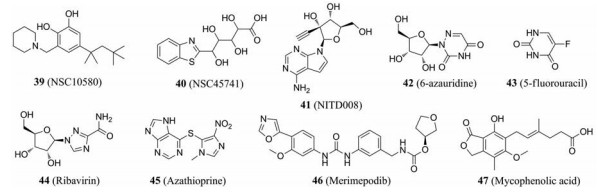
2.1 嘧啶合成抑制剂NITD008 (41)作为一种嘧啶合成抑制剂, 具有较好的体外抗ZIKV病毒活性(EC50 = 0.28~0.95 μmol·L-1)。给药剂量为50 mg·kg-1·d-1时, NITD008对ZIKV感染的A129小鼠具有抗病毒活性。此外, 存活的小鼠没有出现任何神经症状[69]。然而, 由于NITD008在临床前评估中展现出导致细胞线粒体减少的毒性作用, 故其应用受到影响[70]。
Pascoalino等[71]以ZIKV感染的Huh 7细胞为模型, 从FDA批准的725种药物(筛选浓度20 μmol·L-1)中筛选出嘧啶合成抑制剂氮尿苷(42, 6-azauridine, EC50 = 2.3 μmol·L-1)和5-氟脲嘧啶(43, 5-fluorouracil, EC50 = 14.3 μmol·L-1)。这两种化合物可抑制胸腺嘧啶核苷酸合成酶(负责催化胸腺嘧啶核苷酸最后一步反应)。但是, 它们被FDA归类为D级妊娠药物[72], 会影响胎儿的正常发育, 故孕妇慎用。
2.2 嘌呤合成抑制剂利巴韦林(44, ribavirin)是一种鸟苷类似物, 对多种RNA和DNA病毒具有广谱活性[73, 74]。其作用机制可分为间接机制和直接机制, 间接机制如次黄嘌呤单磷酸脱氢酶(IMPDH)抑制和免疫调节作用; 直接机制如RNA加帽干扰、聚合酶抑制和致死突变等[74]。利巴韦林通常用于HCV感染的联合治疗。已有研究证实, 利巴韦林对在不同宿主细胞中的不同地理来源的ZIKV株均有抑制作用, 如Vero细胞、人神经祖细胞(hNPCs)、人真皮成纤维细胞(HDFS)和人肺腺癌细胞(A549)[31, 75]。此外, 利巴韦林还能抑制ZIKV感染的STAT-1缺陷小鼠的病毒血症[63]。
硫唑嘌呤(45, azathioprine)是一种嘌呤合成抑制剂(也具有免疫抑制作用), 已被证明可以消除ZIKV在HeLa细胞和JEG3细胞中的复制, 但是不建议在孕妇中使用[72]。
2.3 IMPDH抑制剂最近的研究表明merimepodib (46, MMPD或VX-497, EC50 = 0.6 μmol·L-1)和麦考酚酸(47, mycophenolic acid, EC50 = 0.08 μmol·L-1, MPA)作为IMPDH抑制剂, 可抑制ZIKV-RNA在不同细胞类型中的复制, 包括Huh-7细胞、人宫颈胎盘细胞、神经干细胞和原代羊膜细胞[72, 76, 77]。
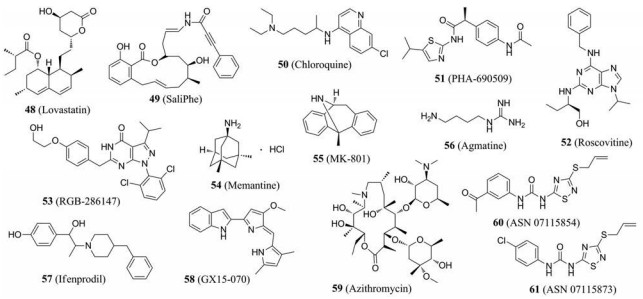
2.4 HMG-CoA还原酶抑制剂Pascoalino等[71]通过剂量效应试验证实了3-羟基-3-甲基戊二酸单酰辅酶A (HMG-CoA)还原酶抑制剂洛伐他汀(48, lovastatin)具有抗ZIKV活性(EC50 = 20.7 μmol·L-1)。此外, Sarkey等[78]证实了洛伐他汀短期大剂量给药可明显减轻小鼠神经系统损伤, 可用于炎症性周围神经疾病, 如ZIKV感染导致的格林-巴利综合征。
2.5 针对囊泡ATP酶的病毒侵入阻断剂Saliphenylhalamide (49, SaliPhe)是一种针对囊泡ATP酶的病毒侵入阻断剂, 并可阻止核内体酸化, 抑制人视网膜色素上皮细胞(RPE)中ZIKV的复制(EC50 = 1 μmol·L-1)[79]。
2.6 病毒复制pH依赖步骤抑制剂氯喹(50, chloroquine)是一种临床应用的经典抗疟药物。该药通过抑制病毒复制过程中的pH依赖步骤来达到抗病毒作用, 其在Vero细胞(EC50 = 9.82 μmol·L-1)、人脑微血管内皮细胞(hBMECs, EC50 = 14.2 μmol·L-1)和人神经干细胞(NSCs, EC50 = 12.36 μmol·L-1)中均显示出显著的抗ZIKV活性。此外, 氯喹还能部分逆转ZIKV引起的小鼠神经球形态学病变。在体外, 氯喹可减少ZIKV感染细胞的数量, 抑制ZIKV感染引起的病毒复制和细胞死亡而不产生细胞毒性[80]。
|
PHA-690509 (51)是一种在研的细胞周期依赖性激酶抑制剂, Xu等[58]筛选发现该化合物抑制ZIKV感染的EC50为1.72 μmol·L-1。该课题组还利用人类神经细胞测定了6 000多种已批准的药物和候选化合物对ZIKV诱导的caspase-3活性和细胞活力的影响, 发现一些小分子可以通过抑制ZIKV复制来保护多种神经细胞避免死亡, 其中, Roscovitine (52, 嘌呤类似物, EC50 = 24 nmol·L-1)和RGB-286147 (53, EC50 = 27 nmol·L-1)在活性较高[58]。
2.8 神经细胞死亡抑制剂Costa等[81]的研究表明ZIKV对中枢神经系统(CNS)具有噬性, 在神经元中优先复制, 诱发神经变性、神经炎症和眼部疾病。ZIKV引发的神经变性可能是由于谷氨酸盐的兴奋毒性所致。FDA批准用于治疗阿尔茨海默病的N-甲基-D-天冬氨酸受体(NMDAR)拮抗药物在体外实验中可以保护ZIKV引起的神经细胞免于死亡, 但未降低病毒滴度, 此类药物包括美金刚胺(54, memantine)、MK-801 (55)、胍丁胺(56, agmatine)和艾芬地尔(57, ifenprodil)等。阻断NMDAR的超活化能降低寨卡病毒引起的细胞死亡, 并有助于改善感染时的神经元症状。Costa等发现美金刚胺在预防ZIKV引起的IFN-α/β R-/-小鼠神经细胞死亡和神经退行性变方面非常有效。此外, 美金刚胺也被FDA列入B类妊娠药物, 因此它可以安全地应用于减轻与ZIKV感染相关的神经并发症。
2.9 Bcl-2蛋白抑制剂Obatoclax mesylate (58, GX15-070), 是一种在研的广谱抗癌药物。属于Bcl-2蛋白家族抑制剂, 它以细胞Mcl-1为靶点, 抑制细胞内吞, 从而诱导细胞凋亡。研究发现, obatoclax mesylate抑制ZIKV的EC50为0.3 μmol·L-1 [79]。
3 其他药物Retallack等[82]通过测试2 177种上市药物抑制Vero细胞感染ZIKV后72 h死亡的效果, 筛选出抗生素、核苷酸及蛋白质合成抑制剂等活性化合物, 但是其中许多化合物对Vero细胞或U87细胞有毒性, 或被禁用于孕妇。然而, 他们发现大环内酯类抗生素阿奇霉素(59, azithromycin)减轻了ZIKV诱导的神经胶质细胞病变作用以及ZIKV感染对U87细胞的影响(EC50 = 2~3 μmol·L-1)。虽然这种药物抗ZIKV的具体机制未知, 但其在妊娠期间使用通常是安全的[83], 故被认为是预防格林-巴利综合征和小头畸形的新选择[71]。
2018年, Micewicz等[84]通过对约250种化合物进行筛选, 发现了4种活性化合物, 其EC50值均在纳摩尔级。他们对其中的ASN 07115854 (60)进行结构优化, 得到一系列衍生物。其中, ASN 07115873 (61)的活性最高(EC50 = 189.2 pmol·L-1), 并且MTT实验显示其毒性也较低。虽然ASN 07115873作用的具体机制仍未知, 但其有望在进一步优化后成为新型抗ZIKV药物。
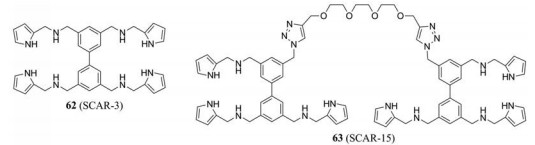
2019年, Kalanidhi等[85]报道了16种具有抗ZIKV活性的合成碳水化合物受体(SCARs), 其机制可能为抑制ZIKV与宿主细胞表面多糖的结合。其中, SCAR 3 (62)和SCAR 15 (63)的活性较高, EC50分别为0.45及0.24 μmol·L-1, 且CC50远大于EC50, 分别为2.43及2.48 μmol·L-1, 具有进一步研究的价值。
4 总结与展望目前, 发现抗寨卡病毒药物的主要途径包括通过结构多样性新化合物库的表型筛选及老药新用, 首先发现高效低毒的先导化合物, 进而通过结构优化与成药性评价, 以得到候选药物。目前文献报道的大多数抗ZIKV活性先导化合物都进行了体外抗病毒活性测试, 部分化合物也在动物模型上进行了体内抗病毒活性测试, 仅有极少的候选化合物进入临床试验。尽管在过去几年中, 研究人员在开发防治ZIKV感染的疫苗和抗病毒药物方面付出了艰苦的努力(表 1), 但是仍没有疫苗或药物批准上市。
|
|
| 表 1 Summary of anti-zika virus agents in this article |
由于宿主细胞在多种病毒生命周期中均扮演重要角色, 故针对宿主细胞研发药物成为了一种颇具吸引力的广谱抗病毒策略。而且, 基于此策略研发出的药物不容易产生耐药性, 因此研发作用于宿主细胞的药物将成为抗寨卡病毒药物重要方向。另外, 鉴于宿主的差异性, 此类药物在研发过程中的动物实验部分应选择与人类亲缘关系较近的模型动物, 以尽量降低进入临床实验后的失败率。
此外, 在开发原创新药的同时, 老药新用理念仍将在未来较长时间内发挥重要作用, 如前文提到的法匹拉韦、西奈芬净和氯硝柳胺, 应继续对老药库进行深入挖掘, 从而尽量缩短药物开发的周期。
不容忽视的是, 源于传统中药的先导化合物筛选与优化是发现抗病毒药物的创新思路。通过加强中药来源的抗寨卡病毒药物原创性研究, 支撑具有我国特色的原创治疗药物的研发。相信随着多种筛选方法及药物设计新策略在抗寨卡病毒药物研发领域的广泛应用, 会有越来越多新颖结构与独特机制的先导化合物被发现, 为治疗ZIKV及相关疾病奠定坚实的基础。
| [1] |
Kuno G, Chang GJ, Tsuchiya KR, et al. Phylogeny of the genus flavivirus[J]. J Virol, 1998, 72: 73-83. DOI:10.1128/JVI.72.1.73-83.1998 |
| [2] |
Dick GW. Zika virus. Ⅱ. Pathogenicity and physical properties[J]. Trans R Soc Trop Med Hyg, 1952, 46: 521-534. DOI:10.1016/0035-9203(52)90043-6 |
| [3] |
Simpson DI. Zika virus infection in man[J]. Trans R Soc Trop Med Hyg, 1964, 58: 335-338. DOI:10.1016/0035-9203(64)90200-7 |
| [4] |
Zanluca C, Melo VC, Mosimann AL, et al. First report of autochthonous transmission of Zika virus in Brazil[J]. Mem Inst Oswaldo Cruz, 2015, 110: 569-572. DOI:10.1590/0074-02760150192 |
| [5] |
Hamelin ME, Baz M, Abed Y, et al. Oseltamivir-resistant pandemic A/H1N1 virus is as virulent as its wild-type counterpart in mice and ferrets[J]. PLoS Pathog, 2010, 6: e1001015. DOI:10.1371/journal.ppat.1001015 |
| [6] |
Chambers TJ, Hahn CS, Galler R, et al. Flavivirus genome organization, expression, and replication[J]. Annu Rev Microbiol, 1990, 44: 649-688. DOI:10.1146/annurev.mi.44.100190.003245 |
| [7] |
Hamel R, Dejarnac O, Wichit S, et al. Biology of Zika virus infection in human skin cells[J]. J Virol, 2015, 89: 8880-8896. |
| [8] |
Van Hemert F, Berkhout B. Nucleotide composition of the Zika virus RNA genome and its codon usage[J]. Virol J, 2016, 13: 95. DOI:10.1186/s12985-016-0551-1 |
| [9] |
Lindenbach BD, Rice CM. Molecular biology of flaviviruses[J]. Adv Virus Res, 2003, 59: 23-61. DOI:10.1016/S0065-3527(03)59002-9 |
| [10] |
Baz M, Boivin G. Antiviral agents in development for Zika virus infections[J]. Pharmaceuticals, 2019, 12: 101. DOI:10.3390/ph12030101 |
| [11] |
Chambers TJ, Weir RC, Grakoui A, et al. Evidence that the N-terminal domain of nonstructural protein NS3 from yellow fever virus is a serine protease responsible for site-specific cleavages in the viral polyprotein[J]. Proc Natl Acad Sci U S A, 1990, 87: 8898-8902. DOI:10.1073/pnas.87.22.8898 |
| [12] |
Wengler G, Czaya G, Farber PM, et al. In vitro synthesis of West Nile virus proteins indicates that the amino-terminal segment of the NS3 protein contains the active centre of the protease which cleaves the viral polyprotein after multiple basic amino acids[J]. J Gen Virol, 1991, 72: 851-858. |
| [13] |
Li H, Clum S, You S, et al. The serine protease and RNA-stimulated nucleoside triphosphatase and RNA helicase functional domains of dengue virus type 2 NS3 converge within a region of 20 amino acids[J]. J Virol, 1999, 73: 3108-3116. DOI:10.1128/JVI.73.4.3108-3116.1999 |
| [14] |
Takegami T, Sakamuro D, Furukawa T. Japanese encephalitis virus nonstructural protein NS3 has RNA binding and ATPase activities[J]. Virus Gen, 1995, 9: 105-112. DOI:10.1007/BF01702653 |
| [15] |
Bartelma G, Padmanabhan R. Expression, purification, and characterization of the RNA 5′-triphosphatase activity of dengue virus type 2 nonstructural protein 3[J]. Virology, 2002, 299: 122-132. DOI:10.1006/viro.2002.1504 |
| [16] |
Guyatt KJ, Westaway EG, Khromykh AA. Expression and purification of enzymatically active recombinant RNA-dependent RNA polymerase (NS5) of the flavivirus Kunjin[J]. J Virol Methods, 2001, 92: 37-44. DOI:10.1016/S0166-0934(00)00270-6 |
| [17] |
Xie X, Gayen S, Kang C, et al. Membrane topology and function of dengue virus NS2A protein[J]. J Virol, 2013, 87: 4609-4622. DOI:10.1128/JVI.02424-12 |
| [18] |
Erbel P, Schiering N, D'Arcy A, et al. Structural basis for the activation of flaviviral NS3 proteases from dengue and West Nile virus[J]. Nat Struct Mol Biol, 2006, 13: 372-373. DOI:10.1038/nsmb1073 |
| [19] |
Munoz-Jordan JL, Sanchez-Burgos GG, Laurent-Rolle M, et al. Inhibition of interferon signaling by dengue virus[J]. Proc Natl Acad Sci USA, 2003, 100: 14333-14338. DOI:10.1073/pnas.2335168100 |
| [20] |
Roosendaal J, Westaway EG, Khromykh A, et al. Regulated cleavages at the West Nile virus NS4A-2K-NS4B junctions play a major role in rearranging cytoplasmic membranes and Golgi trafficking of the NS4A protein[J]. J Virol, 2006, 80: 4623-4632. |
| [21] |
Mackenzie JM, Jones MK, Young PR. Immunolocalization of the dengue virus nonstructural glycoprotein NS1 suggests a role in viral RNA replication[J]. Virology, 1996, 220: 232-240. DOI:10.1006/viro.1996.0307 |
| [22] |
Batista MN, Braga ACS, Campos GRF, et al. Natural products isolated from oriental medicinal herbs inactivate Zika virus[J]. Viruses, 2019, 11: 49. DOI:10.3390/v11010049 |
| [23] |
Haddad JG, Koishi AC, Gaudry A, et al. Doratoxylon apetalum, an indigenous medicinal plant from Mascarene Islands, is a potent inhibitor of Zika and Dengue virus infection in human cells[J]. Int J Mol Sci, 2019, 20: 2382. DOI:10.3390/ijms20102382 |
| [24] |
Tychan Pte Ltd. Safety and Tolerability of an Antibody Against Zika Virus (Tyzivumab) in ZIKV Infected Patients. Clinical Trials. gov.NCT number: NCT03776695. 2018 Dec. NCT03776695. Available online: https://clinicaltrials.gov/ct2/show/NCT03776695 (accessed on 30 May 2019).
|
| [25] |
Niu X, Zhao L, Qu L, et al. Convalescent patient-derived monoclonal antibodies targeting different epitopes of E protein confer protection against Zika virus in a neonatal mouse model[J]. Emerg Microb Infect, 2019, 8: 749-759. DOI:10.1080/22221751.2019.1614885 |
| [26] |
Duan W, Song H, Wang H, et al. The crystal structure of Zika virus NS5 reveals conserved drug targets[J]. EMBO J, 2017, 36: 919-933. DOI:10.15252/embj.201696241 |
| [27] |
Eyer L, Nencka R, Huvarova I, et al. Nucleoside inhibitors of Zika virus[J]. J Infect Dis, 2016, 214: 707-711. DOI:10.1093/infdis/jiw226 |
| [28] |
Arnold JJ, Sharma SD, Feng JY, et al. Sensitivity of mitochondrial transcription and resistance of RNA polymerase II dependent nuclear transcription to antiviral ribonucleosides[J]. PLoS Pathog, 2012, 8: e1003030. DOI:10.1371/journal.ppat.1003030 |
| [29] |
Chen YL, Yin Z, Lakshminarayana SB, et al. Inhibition of dengue virus by an ester prodrug of an adenosine analog[J]. Antimicrob Agents Chemother, 2010, 54: 3255-3261. DOI:10.1128/AAC.00397-10 |
| [30] |
Furuta Y, Takahashi K, Shiraki K, et al. T-705 (favipiravir) and related compounds: novel broad-spectrum inhibitors of RNA viral infections[J]. Antiviral Res, 2009, 82: 95-102. DOI:10.1016/j.antiviral.2009.02.198 |
| [31] |
Baz M, Goyette N, Griffin BD, et al. In vitro susceptibility of geographically and temporally distinct Zika viruses to favipiravir and ribavirin[J]. Antivir Ther, 2017, 22: 613-618. DOI:10.3851/IMP3180 |
| [32] |
Cai L, Sun Y, Song Y, et al. Viral polymerase inhibitors T-705 and T-1105 are potential inhibitors of Zika virus replication[J]. Arch Virol, 2017, 162: 2847-2853. DOI:10.1007/s00705-017-3436-8 |
| [33] |
Delang L, Abdelnabi R, Neyts J. Favipiravir as a potential countermeasure against neglected and emerging RNA viruses[J]. Antiviral Res, 2018, 153: 85-94. DOI:10.1016/j.antiviral.2018.03.003 |
| [34] |
Reznik SE, Ashby CR Jr. Sofosbuvir: an antiviral drug with potential efficacy against Zika infection[J]. Int J Infect Dis, 2017, 55: 29-30. DOI:10.1016/j.ijid.2016.12.011 |
| [35] |
Bullard-Feibelman KM, Govero J, Zhu Z, et al. The FDA-approved drug sofosbuvir inhibits Zika virus infection[J]. Antiviral Res, 2017, 137: 134-140. DOI:10.1016/j.antiviral.2016.11.023 |
| [36] |
Sacramento CQ, de Melo GR, de Freitas CS, et al. The clinically approved antiviral drug sofosbuvir inhibits Zika virus replication[J]. Sci Rep, 2017, 7: 40920. DOI:10.1038/srep40920 |
| [37] |
Perales C, Domingo E. Antiviral strategies based on lethal mutagenesis and error threshold[J]. Curr Top Microbiol Immunol, 2016, 392: 323-339. |
| [38] |
Lazear HM, Govero J, Smith AM, et al. A mouse model of Zika virus pathogenesis[J]. Cell Host Microbe, 2016, 19: 720-730. DOI:10.1016/j.chom.2016.03.010 |
| [39] |
Mangia A, Piazzolla V. Overall efficacy and safety results of sofosbuvir-based therapies in phase Ⅱ and Ⅲ studies[J]. Dig Liver Dis, 2014, 46: 179-185. DOI:10.1016/j.dld.2014.09.026 |
| [40] |
Eyer L, Zouharova D, Sirmarova J, et al. Antiviral activity of the adenosine analogue BCX4430 against West Nile virus and tick-born eflaviviruses[J]. Antiviral Res, 2017, 142: 63-67. DOI:10.1016/j.antiviral.2017.03.012 |
| [41] |
Warren TK, Wells J, Panchal RG, et al. Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430[J]. Nature, 2014, 508: 402-405. DOI:10.1038/nature13027 |
| [42] |
Julander JG, Siddharthan V, Evans J, et al. Efficacy of the broad-spectrum antiviral compound BCX4430 against Zika virus in cell culture and in a mouse model[J]. Antiviral Res, 2017, 137: 14-22. DOI:10.1016/j.antiviral.2016.11.003 |
| [43] |
Coutard B, Barral K, Lichière J, et al. Zika virus methyltransferase: structure and functions for drug design perspectives[J]. J Virol, 2017, 91: e02202-16. DOI:10.1128/JVI.02202-16 |
| [44] |
Zhang CY, Feng TT, Cheng JB, et al. Structure of the NS5 methyltransferase from Zika virus and implications in inhibitor design[J]. Biochem Biophys Res Commun, 2017, 492: 624-630. |
| [45] |
Hamil RL, Hoehn MM. A9145, a new adenine-containing antifungal antibiotic. I. Discovery and isolation[J]. J Antibiot Tokyo, 1973, 26: 463-465. DOI:10.7164/antibiotics.26.463 |
| [46] |
Hercik K, Brynda J, Nencka R, et al. Structural basis of Zika virus methyltransferase inhibition by sinefungin[J]. Arch Virol, 2017, 162: 2091-2096. |
| [47] |
Zhang J, Zheng YG. SAM/SAH analogs as versatile tools for SAM-dependent methyltransferases[J]. ACS Chem Biol, 2016, 11: 583-597. DOI:10.1021/acschembio.5b00812 |
| [48] |
Robert-Gero M, Lawrence F, Lederer E. Potential Clinical Use of Sinefungin: Reduction of Toxicity and Enhancement of Activity[M]. Leishmaniasis, Boston: Springer, 1989: 79-883.
|
| [49] |
Zhang Z, Li Y, Loh YR, et al. Crystal structure of unlinked NS2B-NS3 protease from Zika virus[J]. Science, 2016, 354: 1597-1600. DOI:10.1126/science.aai9309 |
| [50] |
Nitsche C, Zhang L, Weigel LF, et al. Peptide-boronic acid inhibitors of flaviviral proteases: medicinal chemistry and structural biology[J]. J Med Chem, 2017, 60: 511-516. DOI:10.1021/acs.jmedchem.6b01021 |
| [51] |
Lei J, Hansen G, Nitsche C, et al. Crystal structure of Zika virus NS2B-NS3 protease in complex with a boronate inhibitor[J]. Science, 2016, 353: 503-505. DOI:10.1126/science.aag2419 |
| [52] |
Lee H, Ren J, Nocadello S, et al. Identification of novel small molecule inhibitors against NS2B/NS3 serine protease from Zika virus[J]. Antiviral Res, 2017, 139: 49-58. DOI:10.1016/j.antiviral.2016.12.016 |
| [53] |
Li Y, Zhang Z, Phoo WW, et al. Structural dynamics of Zika virus NS2B-NS3 protease binding to dipeptide inhibitors[J]. Structure, 2017, 25: 1242-1250. DOI:10.1016/j.str.2017.06.006 |
| [54] |
Li Y, Zhang Z, Phoo WW, et al. Structural insights into the inhibition of Zika virus NS2B-NS3 protease by a small-molecule inhibitor[J]. Structure, 2018, 26: 555-564. DOI:10.1016/j.str.2018.02.005 |
| [55] |
Roy A, Lim L, Srivastava S, et al. Solution conformations of Zika NS2B-NS3pro and its inhibitionby natural products from edible plants[J]. PLoS One, 2017, 12: e0180632. |
| [56] |
Li Z, Brecher M, Deng YQ, et al. Existing drugs as broad-spectrum and potent inhibitors for Zika virus by targeting NS2B-NS3 interaction[J]. Cell Res, 2017, 27: 1046-1064. DOI:10.1038/cr.2017.88 |
| [57] |
Jurgeit A, McDowell R, Moese S, et al. Niclosamide is a protoncarrier and targets acidic endosomes with broad antiviral effects[J]. PLoS Pathog, 2012, 8: e1002976. DOI:10.1371/journal.ppat.1002976 |
| [58] |
Xu M, Lee EM, Wen Z, et al. Identification of small-molecule inhibitors of Zika virus infection and induced neural cell death via a drug repurposing screen[J]. Nat Med, 2016, 22: 1101-1107. |
| [59] |
Yuan S, Chan JF, den-Haan H, et al. Structure-based discovery of clinically approved drugs as Zika virus NS2B-NS3 protease inhibitors that potently inhibit Zika virus infection in vitro and in vivo[J]. Antiviral Res, 2017, 145: 33-43. DOI:10.1016/j.antiviral.2017.07.007 |
| [60] |
Phoo WW, Zhang Z, Wirawan M, et al. Structures of Zika virus NS2B-NS3 protease in complex with peptidomimetic inhibitors[J]. Antiviral Res, 2018, 160: 17-24. DOI:10.1016/j.antiviral.2018.10.006 |
| [61] |
Yao Y, Huo T, Lin YL, et al. Discovery, X-ray crystallography and antiviral activity of allosteric inhibitors of flavivirus NS2B-NS3 protease[J]. J Am Chem Soc, 2019, 141: 6832-6836. DOI:10.1021/jacs.9b02505 |
| [62] |
Jain R, Coloma J, García-Sastre A, et al. Structure of the NS3 helicase from Zika virus[J]. Nat Struct Mol Biol, 2016, 23: 752-754. DOI:10.1038/nsmb.3258 |
| [63] |
Aguilar JS, Rice M, Wagner EK. The polysulfonated compound suramin blocks adsorption and lateraldifusion of herpes simplex virus type-1 in Vero cells[J]. Virology, 1999, 258: 141-151. |
| [64] |
Baba M, Konno K, Shigeta S, et al. Selective inhibition of human cytomegalovirus replication by naphthalene disulfonic acid derivatives[J]. Antiviral Res, 1993, 20: 223-233. DOI:10.1016/0166-3542(93)90022-B |
| [65] |
Ellenbecker M, Lanchy JM, Lodmell JS. Inhibition of Rift Valley fever virus replication and perturbation of nucleocapsid-RNA interactions by suramin[J]. Antimicrob Agents Chemother, 2014, 58: 7405-7415. DOI:10.1128/AAC.03595-14 |
| [66] |
Wang Y, Qing J, Sun Y, et al. Suramin inhibits EV71 infection[J]. Antiviral Res, 2014, 103: 1-6. DOI:10.1016/j.antiviral.2013.12.008 |
| [67] |
Albulescu IC, Kovacikova K, Tas A, et al. Suramin inhibits Zika virus replication by interfering with virus attachment and release of infectious particles[J]. Antiviral Res, 2017, 143: 230-236. DOI:10.1016/j.antiviral.2017.04.016 |
| [68] |
Zhu S, Zhang C, Huang LS, et al. Discovery and computational analyses of novel small molecule Zika virus inhibitors[J]. Molecules, 2019, 24: 1465. DOI:10.3390/molecules24081465 |
| [69] |
Deng YQ, Zhang NN, Li CF, et al. Adenosine analog NITD008 is a potent inhibitor of Zika virus[J]. Open Forum Infect Dis, 2016, 3: ofw175. DOI:10.1093/ofid/ofw175 |
| [70] |
Munjal A, Khandia R, Dhama K, et al. Advances in developing therapies to combat Zika virus: current knowledge and future perspectives[J]. Front Microbiol, 2017, 8: 1469. |
| [71] |
Pascoalino BS, Courtemanche G, Cordeiro MT, et al. Zika antiviral chemotherapy: identification of drugs and promising starting points for drug discovery from an FDA-approved library[J]. F 1000Res, 2016, 5: 2523. |
| [72] |
Barrows NJ, Campos RK, Powell ST, et al. A screen of FDA-approved drugs for inhibitors of Zika virus infection[J]. Cell Host Microbe, 2016, 20: 259-270. DOI:10.1016/j.chom.2016.07.004 |
| [73] |
Graci JD, Cameron CE. Mechanisms of action of ribavirin against distinct viruses[J]. Rev Med Virol, 2006, 16: 37-48. DOI:10.1002/rmv.483 |
| [74] |
Sidwell RW, Huffman JH, Khare GP, et al. Broad-spectrum antiviral activity of virazole:1-beta-D-ribofuranosyl-1, 2, 4-triazole-3-carboxamide[J]. Science, 1972, 177: 705-706. DOI:10.1126/science.177.4050.705 |
| [75] |
Kamiyama N, Soma R, Hidano S, et al. Ribavirin inhibits Zika virus (ZIKV) replication in vitro and suppresses viremiain ZIKV-infected STAT1-deficient mice[J]. Antiviral Res, 2017, 146: 1-11. DOI:10.1016/j.antiviral.2017.08.007 |
| [76] |
Tong X, Smith J, Bukreyeva N, et al. Merimepodib, an IMPDH inhibitor, suppresses replication of Zika virus and other emerging viral pathogens[J]. Antiviral Res, 2018, 149: 34-40. DOI:10.1016/j.antiviral.2017.11.004 |
| [77] |
Rausch K, Hackett BA, Weinbren NL, et al. Screening bioactives reveals nanchangmycin as a broad spectrum antiviral active against Zika virus[J]. Cell Rep, 2017, 18: 804-815. DOI:10.1016/j.celrep.2016.12.068 |
| [78] |
Sarkey JP, Richards MP, Stubbs EB. Lovastatin attenuates nerve injury in an animal model of Guillain-Barre syndrome[J]. J Neurochem, 2007, 100: 1265-1277. DOI:10.1111/j.1471-4159.2006.04309.x |
| [79] |
Kuivanen S, Bespalov MM, Nandania J, et al. Obatoclax, saliphenylhalamide and gemcitabine inhibit Zika virus infection in vitro and differentially affect cellular signaling, transcription and metabolism[J]. Antiviral Res, 2017, 139: 117-128. |
| [80] |
Delvecchio R, Higa LM, Pezzuto P, et al. Chloroquine, an endocytosis blocking agent, inhibits Zika virus infection in different cell models[J]. Viruses, 2016, 8: 322. |
| [81] |
Costa VV, Del Sarto JL, Rocha RF, et al. N-Methyl-d-aspartate (NMDA) receptor blockade prevents neuronal death induced by Zika virus infection[J]. MBio, 2017, 8: e00350-17. |
| [82] |
Retallack H, Di Lullo E, Arias C, et al. Zika virus cell tropism in the developing human brain and inhibition by azithromycin[J]. Proc Natl Acad Sci U S A, 2016, 113: 14408-14413. DOI:10.1073/pnas.1618029113 |
| [83] |
Lin KJ, Mitchell AA, Yau WP, et al. Safety of macrolides during pregnancy[J]. Am J Obstet Gynecol, 2013, 208: 221. |
| [84] |
Micewicz ED, Khachatoorian R, French SW, et al. Identification of novel small-molecule inhibitors of Zika virus infection[J]. Bioorg Med Chem Lett, 2018, 28: 452-458. DOI:10.1016/j.bmcl.2017.12.019 |
| [85] |
Kalanidhi P, Anjali J, Tugba M, et al. Anti-Zika activity of a library of synthetic carbohydrate receptors[J]. J Med Chem, 2019, 62: 4110-4119. DOI:10.1021/acs.jmedchem.9b00142 |