 2020, Vol. 55
2020, Vol. 55



2. 山东省农业科学院家禽研究所, 山东 济南 250012
2. Institute of Poultry Science, Shandong Academy of Agricultural Sciences, Jinan 250012, China
流感(influenza或者flu)是由流感病毒(influenza virus, IFV)引起的急性上呼吸道疾病。流感病毒属于正黏病毒科(Orthonlyxovoridae), 是一种单股、负链RNA基因组病毒[1], 含有多种亚型, 其中甲型流感病毒传染性较强, 是引起季节性或者大流行流感的主要病毒。一旦高致病性的流感病毒在全球蔓延开来, 将会造成严重的社会健康危机和生命损失。到目前为止, 有文献记载的流感大流行共有5次: ① 1918年西班牙H1N1型流感; ② 1957年亚洲H2N2型流感; ③ 1968年中国香港H3N2型流感; ④ 2005年H1N1型禽流感; ⑤ 2009年H1N1型流感[2-4]。如今, 流感仍然对全球人类的健康存在威胁, 根据最近的一项研究, 季节性流感影响了大约20%的世界人口, 每年可造成25万至50万人死亡[5]。
流感病毒感染宿主细胞后的复制速度非常迅速, 其生命周期大体可分8个环节:吸附(binding); 内吞(endosome); 融合(fusion); 复制(replication); 翻译(translation); 装配(assembly); 出芽(budding)和释放(release) (图 1)[6]。目前, 对抗流感病毒的策略有两种:疫苗和小分子抗流感药物。其中, 接种流感疫苗是预防流感最有效方法, 研制新型、高速、高效、广谱的流感疫苗一直是各国应对流感暴发的主要手段。现在主要接种的流感疫苗是三价灭活疫苗和减毒活疫苗[7, 8]。但疫苗每年都需要重新配置, 以适应抗原变异, 并且其研发周期长, 成本高, 这些缺点使得小分子药物成为防治流感的主要手段。目前上市或处于临床阶段的抗病毒小分子化合物主要包括特异性流感病毒抑制剂(M2离子通道阻滞剂、NA抑制剂、PA抑制剂和PB2抑制剂)和一些广谱抗病毒药物(利巴韦林、硝唑尼特、盐酸阿比多尔、法匹拉韦等)[9-11]。本综述根据流感病毒生命周期中各靶点, 精选近几年研究实例, 从药物化学的角度总结了靶向流感病毒小分子抑制剂的研究进展和现状, 旨在使相关领域研究人员能快速把握流感病毒抑制剂的发展动态并为他们寻找新的靶点或抑制剂提供理论基础。
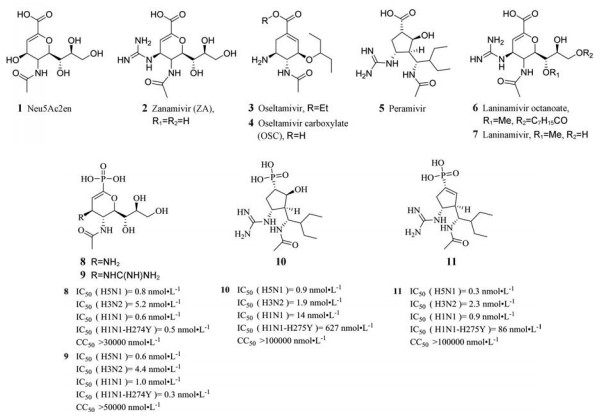
1 神经氨酸酶(NA)抑制剂神经氨酸酶(neuraminidase, NA), 别名唾液酸酶(sialidase), 是嵌入流感病毒包膜表面的一种重要的功能糖蛋白, 由病毒RNA的第6节段负责编码[12, 13]。NA共有11个亚型(N1~N11), 其结构是一个四聚体, 由头部执行生物学催化功能: ①促进子代病毒颗粒从宿主细胞中释放; ②阻止释放后子代病毒颗粒的聚集; ③阻止病毒灭活, 促进病毒在呼吸道中的传播和进入呼吸道上皮细胞, 在流感病毒的生命周期中发挥着关键作用[14]。随着NA和NA与天然底物唾液酸的共晶体结构相继解析[15], 酶活性位点与抑制剂的详细作用机制也随之得到确认, 大大加快了新型神经氨酸酶抑制剂(NAIs)的发现。
目前, 已有4种药物被批准用于临床治疗, 包括扎那米韦(zanamivir, ZAN, Relenza®)、磷酸奥司他韦(oseltamivir phosphate, Tamiflu®)、帕拉米韦(peramivir1, Rapivab®)和辛酸拉尼米韦(laninamiviroctanoata, Inavir®)。
1974年, Palese等[16]分析唾液酸在NA中发生催化反应所产生的过渡态结构, 首次发现了一种唾液酸类似物NA抑制剂Neu5Ac2en (1), 但进一步的活性研究显示, 其体内排泄较快, 对感染小鼠仅有较弱的治疗作用[17]。随着对NA晶体结构研究的进一步深入, 1991年, 扎那米韦(2)研制成功, 并于1999年7月获美国食品和药品管理局(FDA)批准。2是1的类似物, 对大多数甲型乙型流感病毒均有较高抑制效果, 但其口服生物利用度较低, 需要通过鼻腔吸入才能获得较好疗效, 并且其合成较为繁琐, 生产成本较高[18, 19]。紧随其后, 1996年, 奥司他韦(3)研制成功, 其磷酸化形式磷酸奥司他韦(4)于1999年10月获FDA批准, 2001在我国获准上市。相较2、4的口服生物利用度大大提高, 成为第一个口服抗流感药物[20]。4的体内外抗病毒试验数据显示, 该药物对各种流感病毒株的复制和传播过程均有较好疗效, 并且毒性较低, 这使其迅速成为全球抗流感病毒的一线药物, 但该药广泛使用使得对其耐药的病毒株不断出现, 极大影响了疗效[21]。帕拉米韦(5)是具有羧酸、胍基和亲脂性侧链的环戊烷衍生物, 2009年获FDA批准进入研发审批的快速通道, 其在体外和动物模型中具有广谱抗流感病毒活性, 抗病毒机制与4相同, 且疗效和不良反应也相似, 但由于生物利用度低, 给药方式为静脉注射[22, 23]。辛酸拉尼米韦(6)是拉尼米韦(7)的前药, 2010年于日本上市, 用于治疗甲型和乙型流感病毒引起的感染, 并且该药对4的耐药株也有较好抑制作用, 其最大优势是可在肺部停留较长时间, 每周只需吸入一次就可有效的抗季节性流感[19, 23]。

|
Figure 1 Influenza A virus life cycle |
现在临床上已经出现了对这4种药物具有耐药性的各种突变株[18-20, 22], 例如H1N1或H5N1的H274Y亚型和H3N2-E119V亚型流感病毒是最常见的NA突变株, 它们对2敏感而对4耐药, 因此严重限制了口服药物4的临床应用[24-26]。对于H5N1-H274Y突变株NA, H274Y的突变是对4耐药的分子基础, 该突变株中组氨酸残基突变为酪氨酸残基扰乱了Glu276的亚甲基形成的疏水口袋, 从而阻碍配体3与结合口袋发生疏水性结合[27]。因此, 迫切需要开发新一代用于治疗流感的NAIs。
以4种上市药物为先导化合物, 通过各种药物化学策略提高活性与抗耐药性是目前抗流感病毒药物的主要研究方向。Shie等[28]依据生物电子等排体策略发现了扎那米韦(2)的磷酸类似物8和9。相较于2, 它们对H1N1、H1N1-H274Y、H3N2和H5N1的抑酶活性和细胞活性有显著提高, 且细胞毒性仍然很小。分子对接显示:磷酸基团比羧酸基团多一个羟基, 磷酸基团在活性位点能形成更多的氢键, 大大提高了与NA催化中心的结合力, 使2的羧基被磷酸基团取代后活性得到提高。但后续他们再将帕拉米韦(5)的羧酸替换为磷酸时, 发现化合物10的抑酶活性和细胞活性均不如2、4和5, 原因可能是5和10中环戊烷环骨架的柔性太大, 磷酸基的引入限制了优势构象的形成。为了降低环戊烷母环骨架的柔性, 在环戊烷骨架中引入双键得到化合物11。其抑酶活性、细胞活性的确比10提高了很多, 并与2、4和5相当[29]。
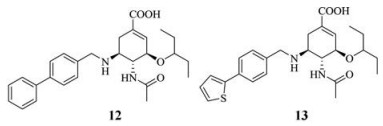
自NA活性中心周围的150腔和430腔相继被发现后, 越来越多的研究开始靶向这两个新位点。2014年, Xie等[30]在4的C-5位氨基上引入取代苄基进行多样性修饰, 得到了两个具有明显N1选择性抑制作用的高活性化合物12和13, 它们对N1 (H5N1-1220)的IC50值分别为1.9和2.1 nmol·L-1; 对N2 (H9N2-S2)的IC50值分别为580和210 nmol·L-1。分子模拟显示12和13的联苯甲基和噻吩联苯甲基可以很准确地嵌入到150腔, 与NA双位点结合的设计思路完全吻合, 通过形成与150腔的附加作用力, 进而提高抑制group-1 NAs的活性。同时该额外作用也补偿了4和其耐药株H5N1-H274Y之间减弱的作用力, 该研究为进一步设计双位点结合型流感病毒NAIs奠定了基础。
|
|
2018年, 本课题组结合上述化合物继续对4进行修饰, 同样运用多位点结合策略, 保持奥司他韦母核及5-位氨基苄基优势取代基不变, 针对150腔位点进行特异性的修饰, 发现14和15对group-1和group-2流感病毒NAs均具有很高的抑制活性, 特别是对09N1、N2、N6和N9亚型的抑制效果, 比4分别高出6.8~12.5倍和1.2~3.9倍(图 2)。它们对N1-H274Y和N2-E119V变异株的抑制活性也均高于4。在细胞水平的抗病毒活性实验中, 它们也表现出了同等或优于4的抗病毒活性。分子动力学模拟研究发现14同时占据了催化中心和150腔, 其4骨架部分与阳性对照4重叠良好, N, N-二乙胺基苯基部分与150腔通过氢键作用、疏水性和范德华力相结合, 使化合物增强了14与NAs的亲和力。初步的成药性评价显示: 14具有较低的体内外毒性(1 g·kg-1的剂量对小鼠灌胃后无毒性表现, CC50 > 200 μmol·L-1)和较高的HLM (t1/2 > 145 min, 60 min remaining: 82.7%)及人血浆(120 min remaining: 120.7%)中的代谢稳定性。此外, 14还表现出较好的体内(鸡胚)抗病毒活性[31]。

|
Figure 2 The structures of compounds 14, 15 |
同时, 为提高奥司他韦(3)衍生物对N1-H274Y突变株的抑制活性, 本课题组基于构效关系并通过取代基的多样性修饰对3进行结构优化, 继续修饰其150腔。活性结果显示:化合物16和17对H5N1、H5N2、H5N6和H5N8表现出了同等或优于3的抗病毒活性, 此外, 在抑酶实验中, 17对N1、N8、N1-H274Y的抑制作用比3高5~86倍, 与2相当。分子动力学(MD)模拟和自由能计算结果表明, 17具有比4更高的NAs亲和力, 主要归因于17与NAs形成的额外范德华力相互作用和形状的互补性, 17除了与催化中心相互作用外, 其二苯基硫醚基团还与150腔之间存在额外相互作用。化合物17在HLM (t1/2 > 145 min, 60 min remaining: 100.6%)和人血浆(120 min remaining: 91.58%)中的代谢稳定性非常高, 在大鼠体内药代动力学测试中也显示出良好的口服生物利用度(10.30%)。另外, 在体内(鸡胚)抗病毒活性实验中, 化合物17抗H5N2型禽流感病毒的活性优于4[32]。
此外, 本课题组还靶向430腔对4的C-1位羧基进行修饰, 发现了一个活性突出的化合物18, 酶和细胞水平上它对H5N1和H5N6亚型毒株的抑制效果与4相当。鸡胚中的抗病毒实验显示, 18对H5N1毒株的敏感性不高, 但在10 mmol·L-1的浓度下, 对H5N6毒株的抑制效果优于4[33]。
2019年, 本课题组基于前期对430腔和150腔化学空间的探索, 以3为先导化合物, 采用分子杂合策略, 通过修饰3的C-1位羧基和C-5位氨基, 设计合成了一系列新型的同时靶向NAs催化中心、150腔和430腔的三位点结合型奥司他韦衍生物。其中, 化合物19活性较好(图 3), 对H5N1和H5N1-H274Y的IC50值为0.044和1.40 μmol·L-1, 均优于4 (IC50分别为0.067和2.15 μmol·L-1), 对H5N1的细胞水平的抗病毒活性EC50值为0.66 μmol·L-1, 也略优于4 (EC50值为0.82 μmol·L-1), 此外, 19在最大测试浓度下(200 μmol·L-1)未显示明显的细胞毒性, 小鼠灌胃给药也进一步验证了19的低急性毒性[34]。

|
Figure 3 Discovery and structure of compound 19 |
M2离子通道在流感病毒感染初期起着重要作用, 该通道是一种同源四聚体的Ⅲ型跨膜蛋白, 由4个M2单位组成, 每个单位含有97个氨基酸残基[35]。M2离子通道的主要功能是转运质子, 诱导病毒膜与内小体膜融合。当受体介导的内吞完成后, M2离子通道会将质子从内吞后形成的内小体中转运到病毒内部, 导致病毒内部酸化, 启动血凝素介导的膜融合, 将病毒核糖核蛋白(vRNP)复合物释放到胞浆中, 启动随后的病毒复制程序。M2离子通道阻滞剂通过与离子通道的内部结合, 阻断质子的流入, 从而阻止血凝素介导的膜融合来发挥作用。
FDA批准的第一批治疗IAV感染的抗病毒药物是金刚烷类, 包括金刚烷胺(20, amantadine)和金刚乙胺(21, rimantadine)。它们可以阻断M2膜蛋白离子通道, 进而阻止M1蛋白与核糖核苷蛋白的解离, 导致甲型流感病毒RNA的复制进程无法启动来达到治疗效果[36, 37]。由于M2蛋白仅存在于甲型流感病毒的膜蛋白中, 故这类药物属于甲型流感病毒特异性抑制剂, 对乙型流感病毒无效, 但此类药物的长期、广泛、大量使用, 导致多数甲型流感病毒产生了严重的耐药性[38, 39]。目前, 在可传播病毒株中发现的3个主要突变是L26F、V27A和S31N, 其中, S31N是95%以上耐药病毒变异位点[40, 41]。此外, 长期服用该类药物可能产生较严重的中枢神经系统不良反应, 因此, 美国疾病预防与控制中心(CDC)不再建议使用M2离子通道阻滞剂来治疗甲型流感[42]。
为解决相应的耐药性问题, 科研人员一直努力通过晶体学、NMR结构分析和分子动力学模拟来研发新型M2通道抑制剂。随着M2蛋白与20和21的多个共晶体结构被解析, 其耐药机制也随之被阐明[43]。目前最多的修饰方法是向金刚烷骨架增添一些附加基团作为抑制剂“弹头”, “弹头”可以有效增大抑制剂的分子体积, 更有效的填充结合位点, 从而增强对野生和突变型M2离子通道阻滞作用[44, 45]。
2011年, Wang等[46]使用计算机辅助设计发现伯胺化合物22对野生型、M2-V27A和M2-L26F突变型均具有良好的抑制活性, 其IC50值分别为18.7、0.3和5.6 μmol·L-1, 抑制活性与20对野生株的抑制活性相当, IC50值为15.7 μmol·L-1。
2014年, Rey-Carrizo等[47]发现了一种具有多环支架结构的吡咯烷衍生物23, 其空间结构与金刚烷胺骨架类似, 通过双电极电压钳法检测证实它具有微摩尔级的野生型M2、M2-V27A和M2-L26F突变型抑制活性, IC50分别为18、0.7和8.6 μmol·L-1, 细胞水平的抗病毒活性测试证明, 化合物23对H1N1的抑制效果(EC50 = 1.8 μmol·L-1)明显优于阳性药物20 (EC50 = 30 μmol·L-1)。
2017年, Barniol-Xicota等[44]根据对金刚烷胺对V27A突变株耐药性机制的阐明, 设计合成了一系列新型金刚烷胺基哌啶化合物, 其中化合物24 (EC50 = 0.14 μmol·L-1)活性最突出, 与20相当(EC50 = 0.14 μmol·L-1)。细胞水平的抗病毒活性测定和双电极电压钳技术测定证实它们为野生型和M2-V27A突变型双重抑制剂。
Li等[48]通过对之前发现的一些S31N抑制剂[41, 49]进行多样性修饰, 在金刚烷胺疏水骨架中加入极性取代基, 增加类药性, 发现化合物25具有亚微摩尔级的抑制活性(EC50 = 0.4 μmol·L-1), 后续又得到一种含有异噁唑取代基的金刚烷胺类化合物26, 对多种甲型流感病毒(M2-S31N突变型、奥司他韦敏感型和耐药型)都具有亚微摩尔级的抑制活性(EC50 = 0.1~0.2 μmol·L-1)[45]。最近, 该团队又对之前活性较好的化合物进行体外药代动力学研究发现了抑制剂27, 其抗病毒活性强, EC50为0.8 μmol·L-1, 体外PK性质良好, 在小鼠和人肝微粒体中的t1/2值均大于145 min, 在Caco-2细胞中具有较高的通透性, 且不抑制CYP的5种亚型, 与奥司他韦也具有较强的协同作用[50]。
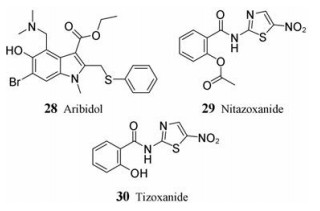
3 血凝素(HA)抑制剂与NA一样, HA也是流感病毒表面一种重要的糖蛋白, 含有多种亚型, 主要功能是识别宿主表面的唾液酸受体, 促进病毒进入宿主细胞。近年来, 靶向HA介导的膜融合新策略获得了众多科研者的关注。在病毒表面, 成熟的HA通常是以同源三聚体形式存在, HA0是HA的前体, 糖基化后裂解形成HA1和HA2, 随后HA2发生构象变化, 导致病毒与目标细胞膜的融合, 释放体内遗传物质, 感染宿主细胞[51]。目前还没有FDA批准上市的HA靶点药物, 但临床研究中前景较好的有阿比多尔(28, aribidol)[52]和硝唑尼特(29, nitazoxanide)[53], 二者分别在美国进行Ⅳ期临床研究和Ⅲ期临床研究。
|
28是一种吲哚骨架化合物, 具有广谱抗病毒活性, 对多种甲型、乙型流感病毒株都具有良好疗效, EC50值为12.9~30.9 μmol·L-1。最近的研究表明, 该药物作用于HA茎部保守区域, 距离融合肽大约16 Å (1Å = 1×10-10 m), 通过与结合位点发生疏水作用而稳定HA, 抑制膜融合。此外, 阿比多尔对多种耐药株都具有敏感性, 并且临床研究期间没有从患者中分离到该抑制剂的耐药株, 是一个非常有开发前景的药物[54]。
2017年, 28作用于血凝素的晶体结构得到解析[54], 依据结构数据, Wright等[55]采用基于结构的药物设计策略, 对28晶体结构附近的一个水分子进行取代, 设计了一系列阿比多尔衍生物, 其中化合物31活性最好(图 4), 亲和力相比先导提高了将近500倍。

|
Figure 4 Structure-based optimization based on the X-ray structure of Arbidol bound to influenza virus hemagglutinin (PDB code 5T6N) |
硝唑尼特是一个前药, 在体内转化为活性形式替唑尼特(30, tizoxanide)发挥作用, 对甲型流感病毒具有广谱的抑制效果, IC50为0.9~3.2 μmol·L-1, 与大多数HA抑制剂的作用机制不同, 硝唑尼特是通过选择性的阻遏病毒血凝素的成熟而在翻译后阶段起作用。同时, 该药物还表现出联合用药作用, 与扎那米韦联用后联合指数(combination index, CI)为0.3~0.48 μmol·L-1, 与奥司他韦联用后联合指数CI为0.18~0.3 μmol·L-1 [56]。
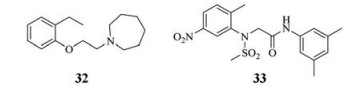
2014年, Basu等[57]运用假病毒的高通量筛选方法(high-throughput screening assay)对一个含有100 000个小分子的化合物库进行筛选, 得到了两个活性突出的抑制剂32和33, 对H1和H5亚型都有良好的抑制效果, IC50值分别为0.3~0.45 μmol·L-1和3.6~5.9 μmol·L-1 [58]。作用机制研究发现二者为流感病毒进入抑制剂, 鸡红血细胞溶血实验(cRBCs)和单克隆抗体竞争实验证明它们结合在HA的茎部区域, 通过抑制HA介导的膜融合而阻止病毒复制。
|
1999年, Plotch等[59]对化合物库进行细胞水平的抗病毒活性筛选, 发现了苗头化合物34 (图 5), IC50为6 μmol·L-1。对其进行进一步结构优化得到化合物35, 抗病毒活性得到大幅度提高, IC50为0.3 μmol·L-1, 细胞水平的抗病毒活性测试结果显示, 35对H5N1毒株的EC50值为27.03 μmol·L-1, 分子模拟和cRBCs实验显示, 化合物35作用于靠近融合肽的HA2茎部保守区域, 以血凝素跨膜亚基HA2介导的膜融合步骤为靶点, 干扰HA的构象变化, 阻碍膜融合[60]。最近, Leiva等[61]对35进行结构优化, 利用氨基取代酰胺基, 同时替换芳香环上不同取代基设计了一系列化合物, 其中化合物36活性最好。细胞水平的抗病毒活性测试显示对H1N1的EC50为4.6 μmol·L-1, HA结合实验和分子动力学模拟证明36作用于HA2茎部区域并与阿比多尔的作用位点部分重叠。

|
Figure 5 Discovery and structural optimization of 34-36 |
2018年, van Dongen等[62]利用一个小蛋白HB80.4[63, 64]模拟CR6261[65]进行竞争性实验, 再用AlphaLISA技术对一个容量约500 000的小分子化合物库进行筛选, 初步选出9 000个活性化合物, 剔除假阳性后, 选出一个苄基哌嗪类苗头化合物37 (图 6), 其抑制H1N1 A/California/07/2009 (H1/Cal)和H5N1 A/Vietnam/1203/2004 (H5/Viet)的IC50值分别是1.39和13.06 μmol·L-1。以37为先导化合物, 在苯基的苄位引入一个酯基, 用苯并噻唑环替换炔基, 发现了38, 对H1到H5亚型流感病毒都有较高的抑制能力, 并且对MDCK细胞没有毒性。但体外药物代谢及药代动力学评价发现, 38的代谢稳定性、溶解度都很差, 而且肝微粒清除率很高, 可能是甲酯和苯并噻唑不稳定所致, 将其替换为苯并噁唑得到39, 肝微粒稳定性有所提高; 随后又用乙酰胺基优化得到四氮唑化合物40, 肝微粒稳定性和溶解度都有较大提升, 体内暴露量也较好, 口服生物利用度约为30%, 动物实验表明, 口服40可以帮助小鼠抵御致命剂量的流感病毒攻击。对不同病毒株进行抗病毒实验, 结果显示, 40选择性地对group 1型病毒抑制, 通过解析40与HA的晶体结构发现, 其作用位点是HA茎部HA1-HA2界面保守区域(图 7), 机制研究表明40通过抑制低pH诱导的HA2构象变化, 阻止病毒释放内部遗传信息, 抑制病毒膜与细胞膜融合来发挥作用[62]。

|
Figure 6 Structural optimization of 37-40 |

|
Figure 7 The binding mode of compound 40 (PDB 6CFG) |
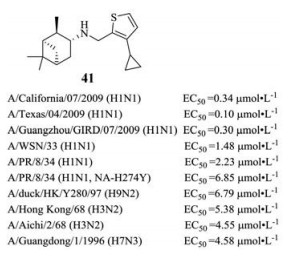
2018年, Zhao等[66]通过对一系列吡那明类(pinanamine)抗病毒药物进行多样性修饰, 发现了一个新颖的HA抑制剂41, 由于其结构与一些报道过的M2-S31N抑制剂非常相似, 最初认为该化合物是通过阻滞M2离子通道来抑制病毒复制, 然而通过将41作为化学探针进行电生理实验探究作用机制时发现, 其真正作用位点是HA2亚基上的一个新的结合口袋, 通过“锁定”HA2的弯曲状态来抑制构象变化发挥作用, 进一步的分子动力学模拟和血凝试验也验证了此结论。这项工作发现了一个新的结合位点并阐述了小分子与HA蛋白相互作用的机制, 表明这一较保守的口袋可能成为抗病毒药物设计和开发的新靶点。
|
流感病毒聚合酶由碱性聚合酶1 (polymerase basic 1, PB1)、碱性聚合酶2 (polymerase basic 2, PB2)和酸性聚合酶(polymerase acidic, PA) 3种蛋白组成。聚合酶复合物“捕获”细胞mRNAs的帽状结构, 并利用它作为启动转录的引物, 进行转录。在这种独特的抓帽过程中, PB2与宿主前mRNA的帽子结合, PA通过其核酸内切酶活性从N-末端5′-帽子处切割约10~14个核苷酸序列, 然后PB1以vRNA作为模板, 连续地将核苷酸添加到裂解后的帽子引物上[67, 68]。此外, 聚合酶复合物还利用互补RNA (cRNA)作为复制中间体进行引物独立合成。流感病毒聚合酶三聚体在病毒复制过程中发挥了关键作用, 成为抗流感病毒药物设计的研究热点。
核苷碱基类似物法匹拉韦(42, Favipiravir, T-705)是一种针对RNA依赖性RAN聚合酶(RdRp)的抗病毒药物, 其抗病毒机制是在人体内转化为三磷酸活性形式发挥作用, 作为流感病毒竞争性聚合酶抑制剂。该药物已被证明对流感病毒株具有广谱抗病毒效果, 包括金刚烷胺和奥司他韦耐药株, 已于2014年在日本批准用于流感治疗, 随后在欧美获批进行临床试验, 目前已进入Ⅲ期临床试验阶段[69]。VX-787 (43)是一种首创的流感病毒PB2抑制剂, 特异性作用于甲型流感, 且适合口服[70]。Ⅱ期临床研究表明, VX-787在甲型流感病毒感染的病毒学和临床评测方面具有非常好的表现[71]。2018年由盐野义制药公司和罗氏共同开发的小分子PA抑制剂Baloxavirmarboxil (44, S-033188)分别于2018年2月和2018年10月先后在日本和美国上市, 商品名Xofluza, 用于治疗甲型和乙型流感, 并对奥司他韦耐药株有效。其疗效显著, 只需要一次服用一片就可以达到抑制流感病毒的效果[72], 最新研究显示I38T突变是病毒对此药产生抗性的主要原因, 而不是之前认为的流感病毒内切酶抑制剂的主要耐药突变E119D[73]。此外, 日本盐野义制药公司开发的另一个PA抑制剂AL-794 (45)也于2016年获批进行Ⅰ期临床研究[70]。
为了发现新颖的强效PA内切酶抑制剂, 2016年, Credille等[74]采用基于片段的药物发现策略(FBDD)对一个含有大约300个分子的金属结合片段库进行筛选, 发现焦袂康酸(pyromeconic acid)骨架是一个对PA有效的配体。随后, 在初期构效关系研究的基础上, 运用片段生长和片段杂合策略对焦袂康酸骨架进行修饰, 最终得到一个抗病毒活性良好的化合物46 (图 8), 其抑制内切酶活性的IC50值为14 nmol·L-1, MDCK细胞水平抗病毒活性EC50为2.1 μmol·L-1, CC50为280 μmol·L-1。

|

|
Figure 8 Fragment-based identification of PA endonuclease inhibitor 46 from a metalloenzyme-focused library |
PB2、PB1和PA蛋白都是病毒聚合酶复合物的组成部分[75], 因此, 干扰蛋白质与蛋白质的相互作用(PPIs)为开发新的抗病毒药物提供了可能。目前, 以PA-PB1相互作用为靶标的PPIs抑制剂研究最为广泛[76]。PB1的N-末端结构域的3个α-螺旋与由PA的C-末端结构域的4个α-螺旋和两个β-发夹结构域形成的口袋会发生相互作用[77], 基于此, Muratore等[78]通过虚拟筛选和ELISA分析测试方法发现了两个对PA-PB1相互作用有干扰的化合物47和48。其中化合物47对多种甲型和乙型流感病毒都具有抗病毒活性, EC50值为12.2~22.5 μmol·L-1。化合物48对A/California/7/9和A/Parma/24/09菌株的作用较弱, EC50分别为75.5和82.2 μmol·L-1。为了提高48的细胞活性, 后续又以化合物48为先导进行结构多样性修饰得到环七噻吩-3-羧酰胺骨架化合物49, 抗病毒效果得到显著增强[79]。2017年, Desantis等[80, 81]继续对环七噻吩-3-羧酰胺骨架进行修饰, 得到化合物50和51, 其对PA-PB1作用较弱, 但细胞水平的抗病毒效果较强, EC50分别为0.18和0.26 μmol·L-1。进一步研究证实51是通过干扰病毒RdRP的两个亚基PA和PB1之间的蛋白质相互作用而抵抗流感病毒, 并且不易导致病毒耐药[82]。
同样的, Tintori等[83, 84]通过高通量对接研究也发现了两个PA-PB1抑制剂52和54, 根据结构对52进一步优化得到化合物53, 其对A/PR/8/34毒株抑制活性EC50为3.5 μmol·L-1, 但这些化合物的体内抗病毒活性未见进一步报道[85]。
2018年, Zhang等[86]运用分子对接对一个小分子化合物库进行筛选, 得到苗头化合物后又运用多组分反应策略进行优化, 最终发现PA-PB1抑制剂55具有广谱抗流感病毒活性, 其中包括金刚烷胺和奥司他韦耐药株, EC50为0.6~2.7 μmol·L-1。值得注意的是, 该化合物在药物选择压力下并未出现耐药性, 值得进一步研究。
除PA-PB1相互作用外, PB1-PB2相互作用也可成为PPIs抑制剂的靶点。2017年, Yuan等[87]采用ELISA筛选方法对大约950个小分子的化合物库进行筛选, 随后对得到的苗头化合物及类似物进行进一步抗病毒筛选, 最后发现一个靶向于PB1-PB2相互作用的小分子抑制剂56, 其EC50为1.4 μmol·L-1, CC50 > 500 μmol·L-1。体外ELISA分析、分子对接以及微型复制子检测方法证实, 化合物56在不同病毒亚型的作用位点都为保守区域, 通过阻断PB1-C端和PB2-N端的相互作用来抑制病毒复制。
5 病毒核蛋白(NP)抑制剂核蛋白(NP)是病毒复制过程中产生的最丰富的病毒蛋白之一。在病毒生命周期中, NP与流感病毒RNA和聚合酶亚基(PB1、PB2和PA)结合, 参与形成病毒核糖核蛋白复合物(vRNPs), 它还涉及vRNPs的核引入、复制和输出[88, 89]。由于NP在病毒复制中的重要作用, 近几年已成为抗病毒药物开发的热点。
萘普生(57, naproxen, 图 9), 一种上市的非甾体抗炎药物, 已被验证可通过抑制NP与RNA的结合来对抗甲型流感病毒, 其体外试验和体内试验对H1N1和H3N2毒株都表现出了良好的抗病毒效果, 在细胞8次传代后也未发现耐药株。表面等离子体共振实验和单点突变实验表明, 57可靶向NP的RNA结合槽, 阻止NP与病毒RNA的相互作用来抑制病毒复制[90, 91]。57及其进一步修饰产物已成为抗炎和抗病毒的双靶点抑制剂[92]。基于此, Tarus等[93]通过片段延伸策略和分子动力学模拟进一步设计了一系列萘普生类似物, 其中化合物58和59对靶点亲和力较为突出, IC50分别为1.1和0.32 μmol·L-1。对其作用位点研究发现, 58和59与结合位点周围氨基酸残基R152和R355形成了两个盐桥, 而萘普生只与R361形成一个盐桥, 这可能是58和59相较于57亲和力增加的原因(图 10)。最近, 该团队又对化合物59做了进一步修饰, 并进行了细胞水平的抗病毒实验, 结果显示衍生物60相较于化合物59活性得到大幅度提升, 机制研究推测, 这些化合物不仅阻碍了NP-RNA的相互作用, 还可以阻碍NP与聚合酶的结合[92]。

|
Figure 9 The structures of compounds 57-60 |

|
Figure 10 Structures of 57-59 bound to nucleoprotein (NP) of influenza A virus |
Nucleozin (61)是一种NP抑制剂, 同时被多个团队通过细胞抗病毒筛选而发现[94, 95]。它可以抑制病毒核蓄积, 在流感病毒生命周期的早期和晚期都有抑制作用, 但在小鼠肝脏微粒体中的水溶性和代谢稳定性较差[88]。Gerritz等[94]以61为先导化合物进行结构修饰发现了两个活性较好的化合物62 [EC50 (H1N1) = 0.04 μmol·L-1, EC50 (H5N1) = 0.07 μmol·L-1]和化合物63 [EC50 (H1N1) = 0.07 μmol·L-1, EC50 (H5N1) = 0.04 μmol·L-1], 其活性相较于先导[EC50 (H1N1) = 0.17 μmol·L-1, EC50 (H5N1) = 0.15 μmol·L-1]提高了1~3倍, 且溶解度和稳定性均明显提高。当剂量超过10 mg·kg-1时, 化合物62能充分保护小鼠免受流感病毒所致的死亡。动态光散射分析和X射线共晶结构的模拟实验证实, 它们的作用机制是抑制NP低聚物的合成来抵抗病毒。
2012年, Cheng等[95]通过骨架跃迁和生物电子等排体策略也设计了一系列Nucleozin类似物, 其中化合物64活性最突出, IC50为0.5~4.6 μmol·L-1, 与Nucleozin活性相当(IC50 = 0.15~3.87 μmol·L-1), 同时对金刚烷胺和奥司他韦耐药株的IC50也低至亚微摩尔级别。
2016年Liao等[96]还发现了一个NP抑制剂65, 与Nucleozin有类似的作用机制, 对H1N1和H3N2的IC50分别为0.27和0.46 μmol·L-1, 小鼠的药代动力学研究表明, 该抑制剂半衰期大于4 h, 生物利用度大于20%, 具备一定类药性。
病毒感染宿主细胞后, 其病毒核蛋白(vRNP)的输出主要是由输出蛋白1 (XPO1)与连接在vRNP的其他核输出蛋白相互作用完成, XPO1的抑制可导致流感病毒复制的减少。基于此, Perwitasari等[97]研制出一个XPO1的选择性抑制剂66, 它对甲型和乙型流感病毒的复制均有较强的抑制效果, 包括大流行的H1N1病毒, 高致病型的H5N1毒株和最近出现的H7N9毒株, EC50都低于微摩尔水平, 药物的小鼠体内试验也证实其对H1N1和H3N2病毒有效。
2015年, Kakisaka等[98]通过对一个含有50 000个分子的化合物库进行抗A/WSN/33 (H1N1)病毒筛选, 发现了一个苗头化合物67, 对多种甲型流感病毒具有良好的抗病毒活性, IC50为0.4~0.63 μmol·L-1。机制研究证明其作用于NP口袋中由RNA结合沟、NP二聚体界面和核输出信号3 (NES3)三部分组成的一个功能区。体内试验证实, 当小鼠腹腔注射67为10 mg·kg-1时, 对感染致死剂量A/WSN/33 (H1N1)的小鼠保护率为25%。但67体内药效低于磷酸奥司他韦, 可能是水溶性差和血浆蛋白结合率过高导致。
2018年, Huang等[99]利用同样的筛选方法对一个含有20 000个化合物的分子库进行筛选, 发现68对H1N1和H3N2病毒株抗病毒效果较好, IC50为0.41~1.41 μmol·L-1。进一步研究证实, 该化合物作用位点与67类似, 是NES3区域以及NP二聚体界面。体内试验结果表明, 68在体内能有效地保护小鼠免受流感病毒感染。最近, Sethy等[100]也筛选出了先导化合物69, 抗甲型流感病毒H1N1的EC50是2.84 μmol·L-1, 选择性指数SI为30, 进行结构修饰得到化合物70, EC50为0.14 μmol·L-1, 选择性指数SI > 785。
White等[101]采用超高通量(ultrahigh throughput screen)筛选方法发现了一个苗头化合物71, 抑制H1N1A/WSN/1933的IC50是0.02 μmol·L-1, 抑制H1N1 A/California/04/2009的IC50为27.43 μmol·L-1。机制研究显示, 71可以诱导NP聚集, 相比在vRNP结构中处于寡聚态的NP, 它对单体NP具有更高亲和力。根据构效关系进行优化得到72, 对甲型和乙型流感病毒都有良好的抑制作用, IC50是1.43~15.15 μmol·L-1, CC50值大于40 μmol·L-1, 与奥司他韦联用还表现出协同抗病毒效果。
6 总结与展望如今, 流感病毒仍然季节性地在人群中肆虐, 高致病型禽流感H5N1和H7N9病毒仍然有人畜共患病的威胁, 但用来对抗流感病毒的“武器”却非常有限。总的来说, 根除流感病毒是不可能的, 因为它具有强大的跨物种传播能力, 尽管新的抗病毒药物不断出现, 但是针对病毒表面蛋白(例如NA和M2)的抑制剂都容易产生耐药性。
目前的抗病毒研究主要分为两个方面, 一是针对现有靶标来优化或者设计新颖的化学实体, 寻找对多种类型的病毒以及耐药株都效果良好的抑制剂, 同时它们也要能抵御自身的耐药突变。目前已经发现针对M2和NA耐药突变体的新的小分子化合物, 这大大鼓励了对已知靶点的有效抑制剂的进一步开发。第二个方面是寻找新的靶标, 开发新的抗病毒药物。近年来药效团模型, 结构生物学和高通量筛选的不断发展加速了靶向病毒聚合酶复合物PA内切酶, PB2帽子结合域和PA-PB1-PB2蛋白-蛋白相互作用靶标的抑制剂发现, 同时宿主细胞内的核转运机制也为调控vRNP复合物的核运输提供新的靶标, 这些靶标在作用方式上都较为独特, 在未来可能有巨大潜力。由聚合酶复合物(PA、PB1、PB2)协同作用产生的独特“抓帽”机制以及它们在不同流感病毒株和亚型上的保守性也为设计新颖的广谱抗病毒抑制剂提供了方向。

|
NP抗原的血清变异性很强, 研究者们曾一度认为NP不适合作为抗流感病毒的药物靶标, 但最近出现的一些NP抑制剂不仅对甲型、乙型流感病毒都有效, 还与奥司他韦展现出了较好的协同作用, 也说明靶向于病毒内部蛋白和外部蛋白的联合治疗能够解决耐药性问题, 几种具有不同机制的抗病毒药物一起使用, 协同发挥作用, 可以获得比单一药物更有效的效果, 同时也增加了毒株的突变障碍。另外, 宿主因子也是一个非常有前景的抗耐药靶标。基因组RNA干扰筛选证实, 许多宿主因子参与流感病毒的复制和转录, 相比靶向病毒蛋白, 靶向宿主因子可以产生突变屏障更高的小分子抑制剂。但这一切还是要注意细胞毒性以及代谢影响, 抗流感病毒研究的最终目标是在不产生耐药突变体的情况下开发安全有效的药物。当前一些具有新结构、新靶标、新机制的化合物已经进入临床试验阶段, 有望在不久的将来为流感病毒感染的防治提供强有力的保障。
| [1] |
Shen Z, Lou K, Wang W. New small-molecule drug design strategies for fighting resistant influenza A[J]. Acta Pharm Sin B, 2015, 5: 419-430. DOI:10.1016/j.apsb.2015.07.006 |
| [2] |
Shin WJ, Seong BL. Novel antiviral drug discovery strategies to tackle drug-resistant mutants of influenza virus strains[J]. Expert Opin Drug Discov, 2019, 14: 153-168. DOI:10.1080/17460441.2019.1560261 |
| [3] |
Peiris JS, Poon LL, Guan Y. Emergence of a novel swine-origin influenza A virus (S-OIV) H1N1 virus in humans[J]. J Clin Virol, 2009, 45: 169-173. DOI:10.1016/j.jcv.2009.06.006 |
| [4] |
Moorthy NS, Poongavanam V, Pratheepa V. Viral M2 ion channel protein: a promising target for anti-influenza drug discovery[J]. Mini Rev Med Chem, 2014, 14: 819-830. |
| [5] |
World Health Organization. Influenza (Seasonal) [DB/OL]. 2018 [2018-11-6]. https://www.who.int/en/news-room/fact-sheets/detail/influenza-(seasonal).
|
| [6] |
Bouvier NM, Palese P. The biology of influenza viruses[J]. Vaccine, 2008, 26: D49-D53. DOI:10.1016/j.vaccine.2008.07.039 |
| [7] |
Taylor DN, Treanor JJ, Sheldon EA, et al. Safety and immunogenicity of a high dosage trivalent influenza vaccine among elderly subjects[J]. Vaccine, 2007, 25: 7656-7663. DOI:10.1016/j.vaccine.2007.08.042 |
| [8] |
Lambert LC, Fauci AS. Influenza vaccines for the future[J]. N Engl J Med, 2010, 363: 2036-2044. DOI:10.1056/NEJMra1002842 |
| [9] |
Goldhill DH, Te Velthuis AJW, Fletcher RA, et al. The mechanism of resistance to favipiravir in influenza[J]. Proc Natl Acad Sci U S A, 2018, 115: 11613-11618. DOI:10.1073/pnas.1811345115 |
| [10] |
Leneva IA, Burtseva EI, Yatsyshina SB, et al. Virus susceptibility and clinical effectiveness of anti-influenza drugs during the 2010-2011 influenza season in Russia[J]. Int J Infect Dis, 2016, 43: 77-84. DOI:10.1016/j.ijid.2016.01.001 |
| [11] |
Haffizulla J, Hartman A, Hoppers M, et al. Effect of nitazoxanide in adults and adolescents with acute uncomplicated influenza: a double-blind, randomised, placebo-controlled, phase 2b/3 trial[J]. Lancet Infect Dis, 2014, 14: 609-618. DOI:10.1016/S1473-3099(14)70717-0 |
| [12] |
Zhang C, Cao YL, Zhong W, et al. Establishment of a cell-based 2009 H1N1 influenza neuraminidase inhibitors evaluation system[J]. Acta Pharm Sin (药学学报), 2010, 45: 383-387. |
| [13] |
Varghese JN, Laver WG, Colman PM, et al. Structure of the influenza virus glycoprotein antigen neuraminidase at 2.9 A resolution[J]. Nature, 1983, 303: 35-40. DOI:10.1038/303035a0 |
| [14] |
McAuley JL, Gilbertson BP, Trifkovic S, et al. Influenza virus neuraminidase structure and functions[J]. Front Microbiol, 2019, 10: 39. DOI:10.3389/fmicb.2019.00039 |
| [15] |
Colman PM, Varghese JN, Laver WG, et al. Structure of the catalytic and antigenic sites in influenza virus neuraminidase[J]. Nature, 1983, 303: 41-44. DOI:10.1038/303041a0 |
| [16] |
Palese P, Schulman JL, Bodo G, et al. Inhibition of influenza and parainfluenza virus replication in tissue culture by 2-deoxy-2, 3-dehydro-N-trifluoroacetylneuraminic acid (FANA)[J]. Virology, 1974, 59: 490-498. DOI:10.1016/0042-6822(74)90458-9 |
| [17] |
Nöhle U, Beau JM, Schauer R. Uptake, metabolism and excretion of orally and intravenously administered, double-labeled N-glycoloylneuraminic acid and single-labeled 2-deoxy-2, 3-dehydro- N-acetylneuraminic acid in mouse and rat[J]. Eur J Biochem, 1982, 126: 543-548. DOI:10.1111/j.1432-1033.1982.tb06815.x |
| [18] |
Von IM, Wu WY, Kok GB, et al. Rational design of potent sialidase-based inhibitors of influenza virus replication[J]. Nature, 1993, 363: 418-423. DOI:10.1038/363418a0 |
| [19] |
Cheer SM, Wagstaff AJ. Zanamivir: an update of its use in influenza[J]. Drugs, 2002, 62: 71-106. |
| [20] |
Mcclellan K, Nz DC, Perry C. Oseltamivir: a review of its use in influenza[J]. Drugs, 2001, 61: 263-283. DOI:10.2165/00003495-200161020-00011 |
| [21] |
Jefferson T, Demicheli V, Rivetti D, et al. Antivirals for influenza in healthy adults: systematic review[J]. Lancet, 2006, 367: 303-313. DOI:10.1016/S0140-6736(06)67970-1 |
| [22] |
Nuttapat A, Watshara S, Tanawut T, et al. Exploring the chemical space of influenza neuraminidase inhibitors[J]. PeerJ, 2016, 4: e1958. DOI:10.7717/peerj.1958 |
| [23] |
Chairat K, Tarning J, White NJ, et al. Pharmacokinetic properties of anti-influenza neuraminidase inhibitors[J]. J Clin Pharmacol, 2013, 53: 119-139. |
| [24] |
Samson M, Pizzorno A, Abed Y, et al. Influenza virus resistance to neuraminidase inhibitors[J]. Antiviral Res, 2013, 98: 174-185. DOI:10.1016/j.antiviral.2013.03.014 |
| [25] |
Memoli MJ, Hrabal RJ, Hassantoufighi A, et al. Rapid selection of oseltamivir- and peramivir-resistant pandemic H1N1 virus during therapy in 2 immunocompromised hosts[J]. Clin Infect Dis, 2010, 50: 1252-1255. DOI:10.1086/651605 |
| [26] |
Dapat C, Kondo H, Dapat IC, et al. Neuraminidase inhibitor susceptibility profile of pandemic and seasonal influenza viruses during the 2009-2010 and 2010-2011 influenza seasons in Japan[J]. Antiviral Res, 2013, 99: 261-269. DOI:10.1016/j.antiviral.2013.06.003 |
| [27] |
Collins PJ, Haire LF, Lin YP, et al. Crystal structures of oseltamivir-resistant influenza virus neuraminidase mutants[J]. Nature, 2008, 453: 1258-1261. DOI:10.1038/nature06956 |
| [28] |
Shie JJ, Fang JM, Lai PT, et al. A practical synthesis of zanamivir phosphonate congeners with potent anti-influenza activity[J]. J Am Chem Soc, 2011, 133: 17959-17965. DOI:10.1021/ja207892q |
| [29] |
Wang PC, Fang JM, Tsai KC, et al. Peramivir phosphonate derivatives as influenza neuraminidase inhibitors[J]. J Med Chem, 2016, 59: 5297-5310. DOI:10.1021/acs.jmedchem.6b00029 |
| [30] |
Xie Y, Xu D, Huang B, et al. Discovery of N-substituted oseltamivir derivatives as potent and selective inhibitors of H5N1 influenza neuraminidase[J]. J Med Chem, 2014, 57: 8445-8458. DOI:10.1021/jm500892k |
| [31] |
Zhang J, Poongavanam V, Kang D, et al. Optimization of N-substituted oseltamivir derivatives as potent inhibitors of group-1 and -2 influenza A neuraminidases, including a drug-resistant variant[J]. J Med Chem, 2018, 61: 6379-6397. DOI:10.1021/acs.jmedchem.8b00929 |
| [32] |
Zhang J, Murugan NA, Tian Y, et al. Structure-based optimization of N-substituted oseltamivir derivatives as potent anti-influenza A virus agents with significantly improved potency against oseltamivir-resistant N1-H274Y variant[J]. J Med Chem, 2018, 61: 9976-9999. DOI:10.1021/acs.jmedchem.8b01065 |
| [33] |
Ju H, Zhang J, Sun Z, et al. Discovery of C-1 modified oseltamivir derivatives as potent influenza neuraminidase inhibitors[J]. Eur J Med Chem, 2018, 146: 220-231. DOI:10.1016/j.ejmech.2018.01.050 |
| [34] |
Jia R, Zhang J, Ai W, et al. Design, synthesis and biological evaluation of "Multi-Site"-binding influenza virus neuraminidase inhibitors[J]. Eur J Med Chem, 2019, 178: 64-80. DOI:10.1016/j.ejmech.2019.05.076 |
| [35] |
Acharya R, Carnevale V, Fiorin G, et al. Structure and mechanism of proton transport through the transmembrane tetrameric M2 protein bundle of the influenza A virus[J]. Proc Natl Acad Sci U S A, 2010, 107: 15075-15080. DOI:10.1073/pnas.1007071107 |
| [36] |
Zhou Z, Liu T, Zhang J, et al. Influenza A virus polymerase: an attractive target for next-generation anti-influenza therapeutics[J]. Drug Discov Today, 2018, 23: 503-518. DOI:10.1016/j.drudis.2018.01.028 |
| [37] |
Kumar B, Asha K, Khanna M, et al. The emerging influenza virus threat: status and new prospects for its therapy and control[J]. Arch Virol, 2018, 163: 831-844. |
| [38] |
Bright RA, Medina MJ, Xu X, et al. Incidence of adamantane resistance among influenza A (H3N2) viruses isolated worldwide from 1994 to 2005: a cause for concern[J]. Lancet, 2005, 366: 1175-1181. DOI:10.1016/S0140-6736(05)67338-2 |
| [39] |
Bright RA, Shay DK, Shu B, et al. Adamantane resistance among influenza A viruses isolated early during the 2005-2006 influenza season in the United States[J]. JAMA, 2006, 295: 891-894. DOI:10.1001/jama.295.8.joc60020 |
| [40] |
Krumbholz A, Schmidtke M, Bergmann S, et al. High prevalence of amantadine resistance among circulating European porcine influenza A viruses[J]. J Gen Virol, 2009, 90: 900-908. DOI:10.1099/vir.2008.007260-0 |
| [41] |
Wang J, Wu Y, Ma C, et al. Structure and inhibition of the drug-resistant S31N mutant of the M2 ion channel of influenza A virus[J]. Proc Natl Acad Sci U S A, 2013, 110: 1315-1320. DOI:10.1073/pnas.1216526110 |
| [42] |
Naesens L, Stevaert A, Vanderlinden E. Antiviral therapies on the horizon for influenza[J]. Curr Opin Pharmacol, 2016, 30: 106-115. DOI:10.1016/j.coph.2016.08.003 |
| [43] |
Thomaston JL, Polizzi NF, Konstantinidi A, et al. Inhibitors of the M2 proton channel engage and disrupt transmembrane networks of hydrogen-bonded waters[J]. J Am Chem Soc, 2018, 140: 15219-15226. DOI:10.1021/jacs.8b06741 |
| [44] |
Barniol-Xicota M, Gazzarrini S, Torres E, et al. Slow but steady wins the race: dissimilarities among new dual inhibitors of the wild-type and the V27A mutant M2 channels of influenza A virus[J]. J Med Chem, 2017, 60: 3727-3738. DOI:10.1021/acs.jmedchem.6b01758 |
| [45] |
Li F, Hu Y, Wang Y, et al. Expeditious lead optimization of isoxazole-containing influenza A virus M2-S31N inhibitors using the Suzuki-Miyaura cross-coupling reaction[J]. J Med Chem, 2017, 60: 1580-1590. DOI:10.1021/acs.jmedchem.6b01852 |
| [46] |
Wang J, Ma C, Fiorin G, et al. Molecular dynamics simulation directed rational design of inhibitors targeting drug-resistant mutants of influenza A virus M2[J]. J Am Chem Soc, 2011, 133: 12834-12841. DOI:10.1021/ja204969m |
| [47] |
Rey-Carrizo M, Barniol-Xicota M, Ma C, et al. Easily accessible polycyclic amines that inhibit the wild-type and amantadine-resistant mutants of the M2 channel of influenza A virus[J]. J Med Chem, 2014, 57: 5738-5747. DOI:10.1021/jm5005804 |
| [48] |
Li F, Ma C, DeGrado WF, et al. Discovery of highly potent inhibitors targeting the predominant drug-resistant S31N mutant of the influenza A virus M2 proton channel[J]. J Med Chem, 2016, 59: 1207-1216. DOI:10.1021/acs.jmedchem.5b01910 |
| [49] |
Wang J, Ma C, Wang J, et al. Discovery of novel dual inhibitors of the wild-type and the most prevalent drug-resistant mutant, S31N, of the M2 proton channel from influenza A virus[J]. J Med Chem, 2013, 56: 2804-2812. DOI:10.1021/jm301538e |
| [50] |
Wang Y, Hu Y, Xu S, et al. In vitro pharmacokinetic optimizations of AM2-S31N channel blockers led to the discovery of slow-binding inhibitors with potent antiviral activity against drug-resistant influenza A viruses[J]. J Med Chem, 2018, 61: 1074-1085. |
| [51] |
Harrison SC. Viral membrane fusion[J]. Nat Struct Mol Biol, 2008, 15: 690-698. DOI:10.1038/nsmb.1456 |
| [52] |
Leneva IA, Falynskova IN, Makhmudova NR, et al. Umifenovir susceptibility monitoring and characterization of influenza viruses isolated during ARBITR clinical study[J]. J Med Virol, 2019, 91: 588-597. DOI:10.1002/jmv.25358 |
| [53] |
Koszalka P, Tilmanis D, Hurt AC. Influenza antivirals currently in late-phase clinical trial[J]. Influenza Other Respir Viruses, 2017, 11: 240-246. DOI:10.1111/irv.12446 |
| [54] |
Kadam RU, Wilson IA. Structural basis of influenza virus fusion inhibition by the antiviral drug Arbidol[J]. Proc Natl Acad Sci U S A, 2017, 114: 206-214. DOI:10.1073/pnas.1617020114 |
| [55] |
Wright ZVF, Wu NC, Kadam RU, et al. Structure-based optimization and synthesis of antiviral drug Arbidol analogues with significantly improved affinity to influenza hemagglutinin[J]. Bioorg Med Chem Lett, 2017, 27: 3744-3748. DOI:10.1016/j.bmcl.2017.06.074 |
| [56] |
Belardo G, Cenciarelli O, La Frazia S, et al. Synergistic effect of nitazoxanide with neuraminidase inhibitors against influenza A viruses in vitro[J]. Antimicrob Agents Chemother, 2015, 59: 1061-1069. DOI:10.1128/AAC.03947-14 |
| [57] |
Basu A, Antanasijevic A, Wang M, et al. New small molecule entry inhibitors targeting hemagglutinin-mediated influenza a virus fusion[J]. J Virol, 2014, 88: 1447-1460. DOI:10.1128/JVI.01225-13 |
| [58] |
Basu A, Komazin-Meredith G, McCarthy C, et al. Molecular mechanism underlying the action of influenza A virus fusion inhibitor MBX2546[J]. ACS Infect Dis, 2017, 3: 330-335. DOI:10.1021/acsinfecdis.6b00194 |
| [59] |
Plotch SJ, O'Hara B, Morin J, et al. Inhibition of influenza A virus replication by compounds interfering with the fusogenic function of the viral hemagglutinin[J]. J Virol, 1999, 73: 140-151. |
| [60] |
Liu S, Li R, Zhang R, et al. CL-385319 inhibits H5N1 avian influenza A virus infection by blocking viral entry[J]. Eur J Pharmacol, 2011, 660: 460-467. DOI:10.1016/j.ejphar.2011.04.013 |
| [61] |
Leiva R, Barniol-Xicota M, Codony S, et al. Aniline-based inhibitors of influenza H1N1 virus acting on hemagglutinin-mediated fusion[J]. J Med Chem, 2018, 61: 98-118. DOI:10.1021/acs.jmedchem.7b00908 |
| [62] |
van Dongen MJP, Kadam RU, Juraszek J, et al. A small-molecule fusion inhibitor of influenza virus is orally active in mice[J]. Science, 2019, 363: eaar6221. DOI:10.1126/science.aar6221 |
| [63] |
Fleishman SJ, Whitehead TA, Ekiert DC, et al. Computational design of proteins targeting the conserved stem region of influenza hemagglutinin[J]. Science, 2011, 332: 816-821. DOI:10.1126/science.1202617 |
| [64] |
Whitehead TA, Chevalier A, Song Y, et al. Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing[J]. Nat Biotechnol, 2012, 30: 543-548. DOI:10.1038/nbt.2214 |
| [65] |
Ekiert DC, Bhabha G, Elsliger MA, et al. Antibody recognition of a highly conserved influenza virus epitope[J]. Science, 2009, 324: 246-251. DOI:10.1126/science.1171491 |
| [66] |
Zhao X, Li R, Zhou Y, et al. Discovery of highly potent pinanamine-based inhibitors against amantadine- and oseltamivir-resistant influenza A viruses[J]. J Med Chem, 2018, 61: 5187-5198. DOI:10.1021/acs.jmedchem.8b00042 |
| [67] |
TeVelthuis AJ, Robb NC, Kapanidis AN, et al. The role of the priming loop in influenza A virus RNA synthesis[J]. Nat Microbiol, 2016, 1: 16029. DOI:10.1038/nmicrobiol.2016.29 |
| [68] |
Reich S, Guilligay D, Pflug A, et al. Structural insight into cap-snatching and RNA synthesis by influenza polymerase[J]. Nature, 2014, 516: 361-366. DOI:10.1038/nature14009 |
| [69] |
Tarbet EB, Maekawa M, Furuta Y, et al. Combinations of favipiravir and peramivir for the treatment of pandemic influenza A/California/04/2009 (H1N1) virus infections in mice[J]. Antiviral Res, 2012, 94: 103-110. DOI:10.1016/j.antiviral.2012.03.001 |
| [70] |
Byrn RA, Jones SM, Bennett HB, et al. Preclinical activity of VX-787, a first-in-class, orally bioavailable inhibitor of the influenza virus polymerase PB2 subunit[J]. Antimicrob Agents Chemother, 2015, 59: 1569-1582. DOI:10.1128/AAC.04623-14 |
| [71] |
U.S.N.L.o. Medicine. A Phase 2b, Randomized, Double-Blind, Placebo Controlled, Parallel-Group, Multicenter Study of 2 Dose Levels of VX 787 Administered as Monotherapy and One Dose Level of VX-787 Administered in Combination With Oseltamivir for the Treatment of Acute Uncomplicated Seasonal Influenza A in Adult Subjects [DB/OL]. 2017 [2017-6-14]. https://www.clinicaltrials.gov/ct2/show/NCT02342249?cond=VX-787&draw=1.
|
| [72] |
U.S.N.L.o. Medicine. A Phase 3, Multicenter, Randomized, Double-blind Study of a Single Dose of S-033188 (Baloxavir Marboxil) Compared With Placebo or Oseltamivir 75 mg Twice Daily for 5 Days in Otherwise Healthy Patients With Influenza [DB/OL]. 2019 [2019-08-22]. https://www.clinicaltrials.gov/ct2/show/NCT02954354?cond=baloxavir.
|
| [73] |
Noshi T, Kitano M, Taniguchi K, et al. In vitro characterization of baloxavir acid, a first-in-class cap-dependent endonuclease inhibitor of the influenza virus polymerase PA subunit[J]. Antiviral Res, 2018, 160: 109-117. DOI:10.1016/j.antiviral.2018.10.008 |
| [74] |
Credille CV, Chen Y, Cohen SM. Fragment-based identification of influenza endonuclease inhibitors[J]. J Med Chem, 2016, 59: 6444-6454. DOI:10.1021/acs.jmedchem.6b00628 |
| [75] |
Boivin S, Cusack S, Ruigrok RW, et al. Influenza A virus polymerase:structural insights into replication and host adaptation mechanisms[J]. J Biol Chem, 2010, 285: 28411-28417. DOI:10.1074/jbc.R110.117531 |
| [76] |
Massari S, Goracci L, Desantis J, et al. Polymerase acidic protein-basic protein 1 (PA-PB1) protein-protein interaction as a target for next-generation anti-influenza therapeutics[J]. J Med Chem, 2016, 59: 7699-7718. DOI:10.1021/acs.jmedchem.5b01474 |
| [77] |
Obayashi E, Yoshida H, Kawai F, et al. The structural basis for an essential subunit interaction in influenza virus RNA polymerase[J]. Nature, 2008, 454: 1127-1131. DOI:10.1038/nature07225 |
| [78] |
Muratore G, Goracci L, Mercorelli B, et al. Small molecule inhibitors of influenza A and B viruses that act by disrupting subunit interactions of the viral polymerase[J]. Proc Natl Acad Sci U S A, 2012, 109: 6247-6252. DOI:10.1073/pnas.1119817109 |
| [79] |
Massari S, Nannetti G, Desantis J, et al. A broad anti-influenza hybrid small molecule that potently disrupts the interaction of polymerase acidic protein-basic protein 1 (PA-PB1) subunits[J]. J Med Chem, 2015, 58: 3830-3842. DOI:10.1021/acs.jmedchem.5b00012 |
| [80] |
Desantis J, Nannetti G, Massari S, et al. Exploring the cycloheptathiophene-3-carboxamide scaffold to disrupt the interactions of the influenza polymerase subunits and obtain potent anti-influenza activity[J]. Eur J Med Chem, 2017, 138: 128-139. DOI:10.1016/j.ejmech.2017.06.015 |
| [81] |
Desantis J, Nannetti G, Massari S, et al. Corrigendum to "Exploring the cycloheptathiophene-3-carboxamide scaffold to disrupt the interactions of the influenza polymerase subunits and obtain potent anti-influenza activity" [Eur. J. Med. Chem. 138 (2017) 128-139][J]. Eur J Med Chem, 2018, 156: 302-303. DOI:10.1016/j.ejmech.2018.07.009 |
| [82] |
Nannetti G, Massari S, Mercorelli B, et al. Potent and broad-spectrum cycloheptathiophene-3-carboxamide compounds that target the PA-PB1 interaction of influenza virus RNA polymerase and possess a high barrier to drug resistance[J]. Antiviral Res, 2019, 165: 55-64. DOI:10.1016/j.antiviral.2019.03.003 |
| [83] |
Tintori C, Laurenzana I, Fallacara AL, et al. High-throughput docking for the identification of new influenza A virus polymerase inhibitors targeting the PA-PB1 protein-protein interaction[J]. Bioorg Med Chem Lett, 2014, 24: 280-282. DOI:10.1016/j.bmcl.2013.11.019 |
| [84] |
Pagano M, Castagnolo D, Bernardini M, et al. The fight against the influenza A virus H1N1: synthesis, molecular modeling, and biological evaluation of benzofurazan derivatives as viral RNA polymerase inhibitors[J]. ChemMedChem, 2014, 9: 129-150. DOI:10.1002/cmdc.201300378 |
| [85] |
Trist IM, Nannetti G, Tintori C, et al. 4, 6-Diphenylpyridines as Promising novel anti-influenza agents targeting the PA-PB1 protein-protein interaction: structure-activity relationships exploration with the aid of molecular modeling[J]. J Med Chem, 2016, 59: 2688-2703. DOI:10.1021/acs.jmedchem.5b01935 |
| [86] |
Zhang J, Hu Y, Foley C, et al. Exploring ugi-azide four-component reaction products for broad-spectrum influenza antivirals with a high genetic barrier to drug resistance[J]. Sci Rep, 2018, 8: 4653. DOI:10.1038/s41598-018-22875-9 |
| [87] |
Yuan S, Chu H, Ye J, et al. Identification of a novel small-molecule compound targeting the influenza A virus polymerase PB1-PB2 interface[J]. Antiviral Res, 2017, 137: 58-66. DOI:10.1016/j.antiviral.2016.11.005 |
| [88] |
Amorim MJ, Read EK, Dalton RM, et al. Nuclear export of influenza A virus mRNAs requires ongoing RNA polymerase II activity[J]. Traffic, 2007, 8: 1-11. DOI:10.1111/j.1600-0854.2006.00507.x |
| [89] |
Cianci C, Gerritz SW, Deminie C, et al. Influenza nucleoprotein: promising target for antiviral chemotherapy[J]. Antivir Chem Chemother, 2012, 23: 77-91. |
| [90] |
Lejal N, Tarus B, Bouguyon E, et al. Structure-based discovery of the novel antiviral properties of naproxen against the nucleoprotein of influenza A virus[J]. Antimicrob Agents Chemother, 2013, 57: 2231-2242. DOI:10.1128/AAC.02335-12 |
| [91] |
Zheng W, Fan W, Zhang S, et al. Naproxen exhibits broad anti-influenza virus activity in mice by impeding viral nucleoprotein nuclear export[J]. Cell Rep, 2019, 27: 1875-1885. DOI:10.1016/j.celrep.2019.04.053 |
| [92] |
Dilly S, FotsoFotso A, Lejal N, et al. From naproxen repurposing to naproxen analogues and their antiviral activity against influenza A virus[J]. J Med Chem, 2018, 61: 7202-7217. DOI:10.1021/acs.jmedchem.8b00557 |
| [93] |
Tarus B, Bertrand H, Zedda G, et al. Structure-based design of novel naproxen derivatives targeting monomeric nucleoprotein of Influenza A virus[J]. J Biomol Struct Dyn, 2015, 33: 1899-1912. |
| [94] |
Gerritz SW, Cianci C, Kim S, et al. Inhibition of influenza virus replication via small molecules that induce the formation of higher-order nucleoprotein oligomers[J]. Proc Natl Acad Sci U S A, 2011, 108: 15366-15371. DOI:10.1073/pnas.1107906108 |
| [95] |
Cheng H, Wan J, Lin MI, et al. Design, synthesis, and in vitro biological evaluation of 1H-1, 2, 3-triazole-4-carboxamide derivatives as new anti-influenza A agents targeting virus nucleoprotein[J]. J Med Chem, 2012, 55: 2144-2153. DOI:10.1021/jm2013503 |
| [96] |
Liao JX, Cheng HM, Wan JT, et al. Evaluation of benzamide derivatives as new influenza A nucleoprotein inhibitors[J]. Open J Med Chem, 2016, 6: 43-50. |
| [97] |
Perwitasari O, Johnson S, Yan X, et al. Verdinexor, a novel selective inhibitor of nuclear export, reduces influenza a virus replication in vitro and in vivo[J]. J Virol, 2014, 88: 10228-10243. |
| [98] |
Kakisaka M, Sasaki Y, Yamada K, et al. A novel antiviral target structure involved in the RNA binding, dimerization, and nuclear export functions of the influenza A virus nucleoprotein[J]. PLoS Pathog, 2015, 11: e1005062. DOI:10.1371/journal.ppat.1005062 |
| [99] |
Huang F, Chen J, Zhang J, et al. Identification of a novel compound targeting the nuclear export of influenza A virus nucleoprotein[J]. J Cell Mol Med, 2018, 22: 1826-1839. DOI:10.1111/jcmm.13467 |
| [100] |
Sethy B, Hsieh CF, Lin TJ, et al. Design, synthesis, and biological evaluation of itaconic acid derivatives as potential anti-influenza agents[J]. J Med Chem, 2019, 62: 2390-2403. DOI:10.1021/acs.jmedchem.8b01683 |
| [101] |
White KM, Abreu P Jr, Wang H, et al. Broad spectrum inhibitor of influenza A and B viruses targeting the viral nucleoprotein[J]. ACS Infect Dis, 2018, 4: 146-157. DOI:10.1021/acsinfecdis.7b00120 |