 2020, Vol. 55
2020, Vol. 55



人巨细胞病毒(human cytomegalovirus, HCMV)是β-疱疹病毒亚家族的成员, 是一种约220 kb的双链DNA病毒。HCMV广泛存在于人类群体中, 易通过直接接触体液(如唾液、眼泪、尿液、粪便、精液和母乳)传播, 在大多数成年人的血清中呈阳性。其血清流行率随地理位置和社会经济条件而变化, 发达国家的血清流行率在30%~70%之间; 在发展中国家, 大多数学龄前儿童(> 90%)都是HCMV抗体阳性[1-3]。对于健康个体, 原发性感染是良性的, 无症状或受自我限制。但对于免疫功能受损人群具有较大的影响, 例如艾滋病患者易患视网膜炎和胃炎, 移植患者易患器官排斥, 新生儿易患大量出生缺陷和发育障碍[4]。据统计, 仅在美国, 每年感染的儿童约40 000名, 其中8 000名发展为长期残疾, 包括小头畸形、宫内生长受限、听力/视力丧失或神经发育迟缓, HCMV感染已成为先天缺陷的主要因素。然而, 目前尚无预防HCMV感染的疫苗, 仅有少数被批准用于治疗HCMV感染的药物, 而且这些药物作用靶点单一、抗病毒活性中等、生物利用度差、易产生耐药性且具有较严重的不良反应[5]。因此, 发现新型高效抗耐药性的抗HCMV药物势在必行。本文根据HCMV的复制周期, 精选近几年的研究实例, 从药物化学角度总结了HCMV抑制剂的研究进展。
1 HCMV的结构HCMV是疱疹病毒科β-疱疹病毒亚家族的典型成员, 是艾滋病、器官移植患者以及出生异常的主要病毒感染源和危及生命的病原体[6]。HCMV与其他疱疹病毒有共同的结构, 都有高度有序的二十面体形状的核衣壳(capsid), 直径约125~130 nm, 包裹着病毒的DNA基因组(dsDNA genome)。核衣壳被一个部分有序的蛋白质层(tegument)包围, 该包被又被包裹在一个多晶型脂质双层中(lipid bilayer envelope), 该多晶型脂质双层含有10多种不同种类的病毒糖蛋白(glycoprotein), 这些糖蛋白负责粘附并进入宿主细胞(图 1)[7]。

|
Figure 1 The structure of HCMV |
HCMV的蛋白质组学研究表明, 其包含100多种自身编码的蛋白质和不完全确定数量的宿主细胞蛋白质。HCMV编码蛋白分布在成熟病毒体衣壳、被膜和包膜3个区域[5, 8]。HCMV的衣壳含有4种独特的非磷酸化蛋白, 大小介于150 kDa (主要衣壳蛋白)到8.5 kDa, 结构类型多, 功能齐全。此外, 还有很多蛋白质在病毒离子的被膜层内磷酸化, 对HCMV的复制至关重要[9]。病毒包膜的组成仍不完全确定, 但包含其他疱疹病毒结构和功能的同系物, 例如gB、gH、gL、gM、gN、gO以及大量特定与β-疱疹病毒和HCMV特异性的蛋白(如gH/gL/UL128/UL130/UL131五聚体复合物), 其中包膜糖蛋白gB和gH/gL形成了病毒进入所需的融合机制。HCMV的自身编码蛋白可改变受感染细胞的细胞免疫反应和脂质代谢, 抑制细胞死亡途径(凋亡和坏死), 促进病毒基因组潜伏期的持久性[10, 11]。这为抗HCMV药物研发提供了重要的靶标。
2015年, Ciferri等[12]在生物化学、功能和结构上对病毒表面的gH/gL、gH/gL/gO和五聚体进行了表征(图 2), 指出了gH/gL/gO和五聚体的细胞特异性入口受体的存在。经SDS/PAGE分析表明, gO和UL128分别与gL形成共价相互作用, 并且五聚体中的UL128-cys162和gH/gL/gO中的gO-cys351与相同的半胱氨酸gL-Cys144形成二硫键。进一步研究发现用MF59佐剂五聚体蛋白免疫的小鼠可提高中和抗体滴度。此外, 对免疫小鼠的血清分析表明, 五聚体疫苗可提高抗体与gH/gL和ULs之间的结合力, 其中和ULs的结合对在上皮细胞和内皮细胞中的HCMV感染具有中和作用。因此针对五聚体的含UL区域的免疫反应的疫苗接种策略具有高效和保护抗体反应的作用, 这对未来研发针对病毒进入的药物具有十分重要的意义。

|
Figure 2 The interaction of gH/gL/gO and pentamer |
与其他疱疹病毒相似, HCMV感染宿主细胞的过程大致可分为吸附与融合、基因组复制、组装与出壳(图 3)[7]。其中HCMV的DNA复制和病毒衣壳的形态变化发生在被感染细胞的细胞核内。聚合的病毒DNA由病毒编码的末端酶切割, 单位长度的线性基因组DNA通过未成熟衣壳的不对称顶点的入口进行包装[13]。离开细胞核的过程中, 包被蛋白的内层被应用于病毒DNA中的衣壳。当粒子沿着细胞骨架成熟时, 被膜蛋白被整合到病毒粒子中, 病毒粒子的最终包裹发生在高尔基远端的细胞内膜室中[14]。HCMV的包膜由宿主衍生的脂质膜组成, 包括20多种病毒编码蛋白, 其中几种病毒膜蛋白已被证明是病毒中和抗体的靶点。获得包膜后, 感染性病毒通过细胞外循环或细胞溶解后释放[15]。

|
Figure 3 The lifecycle of HCMV and the structure at different cleavage stages |
HCMV基因组是人类所有疱疹病毒中的最大基因组(含有235 000个碱基对), 编码大量的保守蛋白以及独特的包膜和被膜蛋白[12]。其基因表达和基因组复制遵循严格调节的级联模式, 即不同类别的病毒基因按不同的顺序来进行, 这是一种典型的疱疹病毒复制策略。首先是即刻早期基因在感染数小时内表达, 其作用是调节宿主细胞对病毒的固有反应并启动转录基因组复制所必须的病毒基因; 然后是早期基因的表达, 其表达产物(DNA聚合酶、末端酶复合物以及磷酸转移酶)可调节宿主细胞功能以促进病毒复制; 最后是晚期病毒基因的表达, 可产生感染病毒离子组装所需的外包裹层和包膜的结构蛋白[16]。目前较多研究是针对基因转录过程中的DNA聚合酶, 今后可深入研究其基因表达过程, 以发现更多的药物作用靶点。
HCMV的复制周期较长, 必须使被感染的细胞存活数天或数周, 以确保成功复制和产生新的感染性病毒颗粒。因此, 细胞存活率对HCMV的持续性感染至关重要。目前文献所记载的HCMV阻断细胞凋亡的途径主要包括: ① HCMV进入靶细胞; ②病毒核衣壳被转运到细胞核, 病毒基因组进入核空间并开始复制, 许多病毒基因产物已被证明可以改变细胞内环境以促进有利于细胞生存的状态; ③即刻早期蛋白IE1和IE2通过AKT磷酸化激活PI(3)K/Akt/mTOR信号通路, 促进感染细胞存活; ④ HCMV蛋白vICA (pUL30)与启动子procaspase-8结合, 阻断其在细胞死亡受体下游的蛋白水解; ⑤ HCMV vMIA (pUL37x1)结合促凋亡的Bax/Bak, 防止线粒体外膜通透性(MOMP)和caspase激活级联的诱导; ⑥ HCMV pUL38可能是通过抑制JNK磷酸化进而抑制内质网(ER)应激诱导的细胞凋亡; ⑦除了描述的病毒蛋白外, HCMV非编码RNAβ2.7可通过与线粒体膜复合体Ⅰ相互作用而阻止细胞凋亡(图 4)[17]。

|
Figure 4 Select HCMV gene products that target apoptic pathways during HCMV infection |
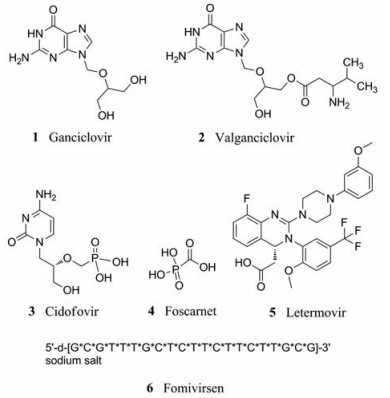
目前已批准6种药物用于治疗HCMV感染, 包括更昔洛韦(ganciclovir, GCV, 1)、缬更昔洛韦(valganciclovir, VGCV, 2)、西多福韦(cidofovir, CDV, 3)、磷甲酸钠(foscarnet, FOS, 4)、勒特莫韦(letermovir, LTV, 5)和福米韦森(formivirsen, 6)。
GCV (1)是一种脱氧尿苷类似物, 是1989年被批准用于治疗HCMV感染的第一种药物, 已成为免疫功能低下者HCMV感染的一线治疗药物[18]。在HCMV感染的细胞中, GCV被病毒蛋白激酶(UL97基因的产物, pUL97)选择性磷酸化, 转化为GCV三磷酸形式, 进而与dGTP竞争, 是HCMV DNA聚合酶有效抑制剂[19]。但由于GCV中的羟基磷酸化而常出现血液学异常, 因此在服用时应根据肾功能适当使用[20, 21]。VGCV (2)是2000年获得批准的GCV的L-缬氨酸酯前药, 口服后在肠壁和肝脏代谢为活性形式(GCV), VGCV的口服生物利用度约为60%。目前, VGCV已经取代了口服GCV, 但对于无症状的高风险移植受者, 预防性治疗还是先发制人治疗仍然存在争议[18]。CDV (3)于1996年被批准用于治疗艾滋病患者的HCMV视网膜炎, 静脉注射给药, 对DNA病毒具有广谱活性。但其肾毒性和口服生物利用度较低, 这限制了其临床效用[22-25]。FOS (4)对多种疱疹病毒均具有体外活性, 被视为二线疗法, 对艾滋病患者的HCMV视网膜炎有效。LTV (5)通过抑制病毒的末端酶复合物, 阻止了DNA到成熟衣壳中的包装。LTV代表一类新型已批准的预防HCMV感染的药物。目前收集的临床数据表明, 移植患者对该药耐受性良好, 有望成为预防HCMV感染的有效疗法[26]。Fomivirsen (6)是一种与编码IE2的mRNA互补的反义磷酸寡核苷酸, 通过眼内注射给药, 于1998年被批准作为二线疗法局部治疗艾滋病患者的HCMV视网膜炎, 其半衰期较长, 但与GCV相比很少使用[27-30]。
4 新型抑制剂研究进展对HCMV复制周期和关键蛋白结构生物学的认识, 为基于靶点的药物设计奠定了良好的基础。组合化学、高通量筛选等技术的运用有力地推动了新型HCMV抑制剂的研究。下面根据病毒复制周期的各个阶段对相关抑制剂进展进行综述, 主要涵盖病毒进入、基因组转录、病毒蛋白质合成和加工、病毒基因组复制以及病毒的成熟与排出。
4.1 DNA聚合酶抑制剂鉴于DNA聚合酶的抗病毒药物研发的成功, 药物化学家对开发针对该酶的抗疱疹病毒药物具有较大的兴趣, 目前这类药物主要包括核苷和核苷酸类似物[31]。
4.1.1 核苷类似物2015年, Sahu等[32]以阿昔洛韦和更昔洛韦为先导化合物, 基于生物各同向性原理设计合成了硒代阿昔洛韦(7)和硒代更昔洛韦(8)。经多种病毒株感染细胞(HEL299 cells)活性测定发现化合物7具有很强的抗HSV-1和HSV-2活性(EC50分别为1.47、6.34 μmol·L-1, CC50 > 100), 而化合物8具有中等的抗HCMV活性(EC50 = 53.1 μmol·L-1, CC50 > 100), 说明硒可作为氧的电子等排体, 这对开发新型核苷类抗病毒药物具有重要启发。
|

|
2017年, Sun等[25]基于前药设计策略设计了4种以N-保护氨基酸或二肽为前体的更昔洛韦单酯前药(图 5), 并进而利用HCMV蛋白酶(HCMVP A143S)筛选实验确定其前体序列, 定量描述组织基质的降解、HCMV蛋白酶的激活以及这些更昔洛韦前药的细胞摄取。研究人员通过采用Michaelis-Menten动力学模拟感染和未感染的细胞环境中前药的水解率, 发现化合物AcPheAbuGCV (9)具有较好的稳定性和细胞摄取率(在血浆中t1/2 = 210 min, 在肝微粒体中t1/2 = 237 min)。这种酯类前药, 可靶向感染患病细胞/组织中的特定酯酶, 同时避免或减少水解。这对已上市药物的优化具有十分重要的参考价值。

|
Figure 5 The discovery of compound 9 |
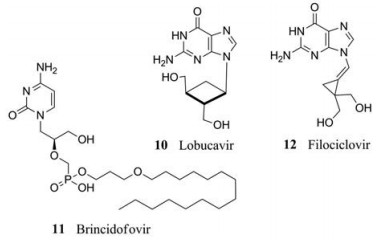
Lobucavir (10)是鸟嘌呤的环丁基类似物, 对大多数疱疹病毒以及HIV和HBV都有活性。LBV是HCMV DNA聚合酶的体外有效抑制剂, 在细胞内磷酸化为三磷酸形式, 但是选择性较差, 在感染细胞中的磷酸化代谢物比未感染细胞仅高出2~3倍[33]。对啮齿动物的毒理学研究表明, 长期服用该药后, 患者所患癌症的种类有所增加。尽管在早期针对HCMV和HBV的临床试验中取得了较好的结果, 但LBV存在安全性问题, 已经停止进一步研发[5]。
Brincidofovir (11)是一种西多福韦的磷酸酯基脂质缀合物。口服时, 其血清浓度高、毒性小。在靶细胞中, 脂质被切割, 游离出西多福韦, 随后被宿主细胞核苷激酶磷酸化, 产生HCMV DNA聚合酶的抑制剂—西多福韦二磷酸。这种修饰基本上消除了肾脏对有机阴离子转运体的吸收, 明显降低了肾毒性, 优于CDV的治疗。在临床研究中, BCV在干细胞移植受者HCMV感染的第二阶段实验中表现良好, 但在第三阶段, 随着时间延长, 产生了一些不良反应(如腹泻), 目前正在将氯噻嗪重新配置成静脉注射药物以避免不良反应[5]。
亚甲基环丙烷类似物是以等位基因类似物为模型而产生, 已被证明具有抗病毒活性。这些类似物的第一代是无环鸟苷的阿昔洛韦生物等位性类似物, 第二代与更昔洛韦的附加羟基密切相关。Zhou等[34, 35]合成了一系列第二代亚甲基环丙烷类似物, 其中2, 2-二羟基甲基衍生物Filociclovir (12)对降低感染HCMV的小鼠死亡率和抑制小鼠内脏器官病毒复制非常有效, 被pUL97磷酸转移酶磷酸化, 进而抑制病毒DNA的合成。Filociclovir具有广谱抗病毒活性, 目前已有有效数据支持FCV进入临床Ⅰ期研究。
4.1.2 核苷酸类似物2017年, Luo等[36]利用前药设计策略扩大了无环核苷磷酸酯[3-氟-2(磷酸单甲氧基)丙基] (FPMPA) (13)的抗病毒谱, 将FPMPA的磷酸与天冬氨酸二酯基的胺基衍生化, 合成了一系列新化合物。其中活性最优的化合物为(S)-FPMPA酰胺前药(14) (图 6), 在低微摩尔浓度下对TZM-BL外周血单核细胞具有抗HIV-1活性, 对HCMV的EC50为0.76 μmol·L-1, CC50为74.70 μmol·L-1。
|

|
Figure 6 The discovery of compound 14 |
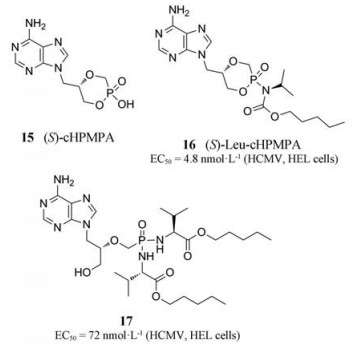
近期, 该课题组进一步基于前药设计策略合成了一系列具有不同氨基酸基序的磷酸酯化(cHPMPA)前药。经过系列活性测定之后, 发现(S)-cHPMPA (15)的磷酰胺酸盐衍生物对疱疹病毒表现出广谱的活性, 其EC50值在低纳摩尔范围内。其中(S)-cHPMPA的亮氨酸酯前药(16)和HPMPA的双磷酰胺前药(17)在人血浆中非常稳定, 半衰期超过1 h, EC50值分别为4.8、72 nmol·L-1 (HEL cells), 并且所有的前药在肝微粒体存在下均发生快速代谢, 具有较小的毒性[37]。这些数据促进了cHPMPA前药的进一步发展, 尤其对于免疫受损的HCMV患者具有十分重要的意义。
此外, 蛋白质-蛋白质的相互作用(protein-protein inhibitors, PPIs)是所有生物过程的核心, 是一类潜在的药物干预治疗靶点。HCMV DNA聚合酶包括一个催化亚单位UL54和一个辅助亚单位UL44, 对DNA复制非常重要。哈佛医学院的研究人员采用等温滴定量热法定量测量了UL54的C末端对应的肽与UL44的结合。结果表明UL54C末端的22个氨基酸肽段与UL44能较好的结合(解离常数为0.7 μmol·L-1)。研究人员进一步通过定点突变实验揭示了两者相互作用界面上的关键氨基酸残基(如Leu1227、Phe1231), 发现任一残基被取代均可极大损害PPI和长链DNA的合成[38]。2016年, Chen等[39]利用高通量筛选发现一个小分子SGM8 (18)以时间依赖性、选择性地抑制UL44和UL54衍生肽的相互作用。其中UL44与SGM8的共晶结构(PDB代码: 5IWD)揭示了SGM8与Lys60残基(图 7橙色虚线)的共价反应以及导致立体冲突的额外非共价相互作用, 这将阻止UL44与UL54的相互作用。综上, 共价结合可作为设计HCMV聚合酶抑制剂的有效策略。
|

|
Figure 7 The structure of SGM8 and main amino acids around the target UL44 of 18 (the orange dotted line represents the covalent interaction) |
US28受体是HCMV双链DNA中编码的病毒G蛋白偶联受体(vGPCRs)之一, 与人类趋化因子受体具有高度同源性, 这些相似性能非常有效地耦合到受感染宿主的信号网络[40]。US28受体具有结合不同的人类趋化因子和CX3C-趋化因子CX3CL1/Fractalkine的能力, 能够在免疫活性宿主中建立终生持续/潜在感染, 并可导致免疫系统不成熟或抑制的患者出现严重和危及生命的疾病。
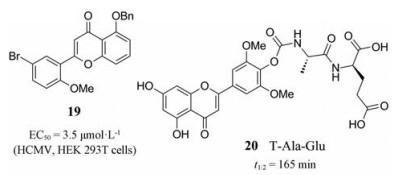
2013年, Kralj等[41]从天然产物黄酮类具有较好抗病毒活性中受到启发, 合成了一系列查耳酮和黄酮类似物, 并对HCMV的US28受体的反向激动剂活性进行筛选。研究人员通过ElK-1报告基因分析发现化合物19对靶标US28的完全反向激动活性为-89%。活性测定得19的EC50值为3.5 μmol·L-1 (HEK 293T cells), 并且对CHO细胞具有较低毒性, 是US28受体信号传导的完全反向激动剂。目前只有两种药物可以抑制US28的组成活性, 化合物19的发现为HCMV药物开发提供了新的结构骨架。
2011年, Ninomiya等[42]利用前药设计策略, 设计合成了系列黄酮氨基酸类衍生物, 并研究了它们的细胞通透性、体外稳定性和体内口服有效性。研究人员利用细胞内各种内源性水解酶对合成的前药进行测试, 发现T-Ala-Glu (20)的半衰期较长(t1/2 = 165 min)。在此基础上, 研究人员进一步测定了20在雄性小鼠中的口服生物利用度, 发现注入之后, 小鼠血浆中的药物浓度明显提高。这些结果表明丙氨酸-谷氨酸结合物是一种潜在的新型抗HCMV药物。
|
即刻早期2蛋白(immediate-early 2 protein, IE2)通过诱导宿主基因表达的广泛失调而在HCMV感染的发病机制中起到直接作用。这种失调导致宿主细胞生理学变化并改变了HCMV诱导的细胞周期改变、免疫调节以及促炎反应的发生[43]。因此, 抑制IE2蛋白表达可以在HCMV感染早期有效地阻断病毒诱导的病理过程。IE2抑制剂的研究对产生耐药性(病毒DNA复制抑制剂)的患者非常重要。由Massari等[44]研制的6-氨基喹诺酮WC5 (21)是目前唯一能够直接阻断IE2蛋白激活的小分子, 能有效地抑制病毒复制(EC50 = 1.0 μmol·L-1), 选择性指数(SI)约为500, 优于GCV。
2013年, 该课题组[44]以WC5为先导化合物, 设计合成了一系列WC5类似物。经过系列活性测试, 发现许多衍生物活性优于GCV, 其中有3种化合物(22~24)的抗病毒活性与先导物WC5相当。综合考虑, 化合物24在人包皮纤维原细胞(human foreskin fibroblast, HFF cells)中的抗HCMV效果最优(EC50 = 1.1 μmol·L-1, CC50 > 500 μmol·L-1, SI > 454) (图 8)。

|
Figure 8 The structures of compounds 21-24 |
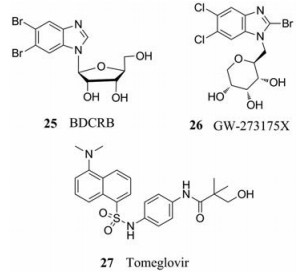
终端酶复合物由几种病毒基因产物组成, 负责将成熟病毒基因组分裂和包装成衣壳, 对衣壳成熟非常重要。在复制过程中, 病毒基因组DNA复制的中间产物形成复杂拓扑结构的多聚体, 终端酶复合物将其切成单位长度的基因组, 并提供将基因组DNA插入未成熟衣壳所需的驱动力。目前已有几种基因产物被证明参与了这一过程, 包括pUL51、pUL52、pUL56、pUL77、pUL89、pUL93和门户蛋白pUL104[5]。迄今为止, 已鉴定出3类终端酶复合物抑制剂, 包括苯并咪唑核苷(benzimidizole ribosides)、多美沙洛韦(tomeglovir)和LTV。
4.4.1 苯并咪唑核苷类化合物(benzimidazole ribosides)BDCRB (25)是最早发现的终端酶抑制剂, 能够阻断门脉蛋白(pUL104)和pUL56的相互作用, 进而阻止DNA插入衣壳[45, 46]。由于8-氧鸟嘌呤DNA糖基化酶和N-甲基嘌呤DNA糖基化酶对BDCRB的快速降解阻碍了其进一步的发展。近期, Uederwood等[47]通过对BDCRB的构效关系研究, 合成了具有相似作用机制且具有较好活性的苯并咪唑核苷GW-273175X (26), 其药代动力学特性得到改善, 蛋白质结合能力降低。
4.4.2 苯二胺磺胺类化合物(phenylenediamine sulphonamides)苯二胺磺胺类化合物是一类新型的非核苷抑制剂, 能特异性阻断HCMV DNA在衣壳中的分裂和包装。Tomeglovir (27)是新型抗HCMV药物, 通过干扰病毒成熟和基因组包装抑制了HCMV的复制[48]。研究表明, 27不仅可以抑制HCMV菌株(包括对现有抗HCMV药物耐药的突变体), 还可以抑制一些猴子和啮齿动物的巨细胞病毒。此外, 文献还报道了27及其主要代谢产物在小鼠、大鼠和犬体内的良好药代动力学数据。尽管有这些有前景的数据, 目前27仍处于临床前开发阶段[49]。
|
2018年, Bongarzone等[1]从人类免疫缺陷病毒药物raltegravir抑制复合物亚蛋白之一UL89的核酸酶功能中受到启发, 以28为先导化合物合成了系列二酮酸类衍生物(α, γ-DKA和β, γ-DKA)。经分子对接确定了化合物29和30与pUL89-C内切酶域活性位点的关键化学区域, 其中二酮酸部分的3个氧原子与活性中心的Mn2+螯合(图 9中的橙色球), 末端苯环与Asn536、Met579和Thr537相互作用, 这种相互作用有助于将抑制剂定位在活性中心的二价金属阳离子附近。图 9定量描述了29上的原子与作用位点之间的距离。经定量UL89核酸酶测定, 发现30能够在低微摩尔范围内在体外抑制UL89 (IC50 = 4.5 μmol·L-1), 是具有开发前景的先导化合物。

|
Figure 9 The structures of compounds 28-30 and the binding mode of 29 and 30 to UL89 |
UL97蛋白激酶是一种丝氨酸/苏氨酸激酶。感染发生后, HCMV病毒会将包括pUL97在内的被膜蛋白释放到宿主细胞中, 以指导病毒蛋白和细胞蛋白的磷酸化以及再分布, 促进成熟病毒粒子的产生[50]。此外, 激酶还可抑制通过CDK抑制剂p21waf1/cip1诱导tip60降解的pul 27, 从而诱导细胞周期停滞[51]。目前UL97蛋白激酶抑制剂主要包括苯并咪唑核苷类、吲哚咔唑类以及喹唑林类3种[52]。
4.5.1 苯并咪唑核苷类化合物(benzimidazolyl nucleosides)Biron等[53]为了改善苯并咪唑核苷的药代动力学, 对其进行构效关系研究时设计合成了maribavir (31), 发现31可抑制UL97激酶和病毒DNA合成, 对CMV和EBV均有抑制作用, 但在体外对其他人疱疹病毒无抑制作用。对HCMV感染的MRC-5肺成纤维细胞进行活性检测, 发现其抗HCMV效价与BDCRB相当, IC50比GCV低4~10倍。31在Ⅰ期临床试验中显示出优良的药代动力学性质, 在大剂量范围内没有中性粒细胞减少或肾毒性; 在Ⅱ期临床试验阶段表现出了较强的抗病毒活性; 目前正在进行Ⅲ期临床试验[24]。
4.5.2 吲哚卡唑类化合物(indole carbazole)Gö6976 (32)、K252c (33)是典型的吲哚咔唑类衍生物, 是对GCV敏感和耐药的HCMV菌株的高效抑制剂, 这类化合物通过降低pUL97蛋白激酶活性来抑制病毒复制, 在HCMV复制周期中表现出长期的抗病毒活性, 与GCV具有拮抗作用[54]。
4.5.3 喹唑啉类化合物(quinazolines)Herget等[55]对一系列喹啉类化合物进行pUL细胞内活性测定, 发现化合物AX7376 (34)、AX7396 (35)和AX7543 (36)在抑制UL97激酶活性并阻断体外底物磷酸化方面具有较高活性, IC50在30~170 nmol·L-1之间。研究人员深入研究其对人成纤维细胞的影响, 发现这些喹啉类化合物可以抑制HCMV复制, EC50值分别为2.4、3.4和3.9 μmol·L-1。这表明喹唑啉是体外靶向pUL97蛋白激酶活性的高效HCMV复制抑制剂。

|
Browne的研究小组[56]通过DNA微阵列试验发现, 在HCMV感染的细胞中, 调节前列腺代谢的基因—环氧合酶(COX-2)上调, 导致前列腺素E2 (PGE2)的合成, 进而干扰了细胞基因表达和免疫功能。随后, Zhu等[57]发现特定的COX-2抑制剂可使HCMV的复制能力下降100倍以上, 这表明COX-2对HCMV的复制至关重要。因此, COX-2抑制剂可使关键病毒调节蛋白的正常水平失衡, 导致病毒复制周期的正常进展在短时间内被阻止。这些研究为抑制HCMV感染提供了新的靶标, 并为识别抗病毒治疗的靶标提供新的策略—微阵列分析。
4.7 以最小衣壳蛋白SCP为靶点HCMV在一个大小相似的衣壳中含有更大的dsDNA基因组, 其密集的基因组会产生较强静电排斥作用, 即需要pp150 (一种仅在巨细胞病毒中发现的包膜蛋白)来稳定其结构。2013年, Dai等[58]借助冷冻电镜(cryoEM)测定了HCMV粒子(含pp150)的亚纳米级的三维结构, 发现pp150由两个螺旋束组成, 这两个螺旋束由一个长的中心螺旋连接。在pp150的一端, 其下螺旋束(lower helix bundle, LHB)与三聚体(Triplex)直接接触(其结合位点如图 10A中的“*”标记), 在另一端, pp150上螺旋束(upper helix bundle, UHB)通过短螺旋(图 10B中的粗箭头所示位置)经SCP (the smallest capsid protein)介导与MCP (the major capsid protein)相互作用。进一步用核酶抑制HCMV感染细胞中SCP的表达会损害含有DNA的病毒颗粒的形成, 并将病毒产量降低到一万分之一。此项研究为治疗HCMV感染提供了一个潜在靶标, 即HCMV-SCP。

|
Figure 10 SCP mediates pp150 binding to the capsid. (A) The model shows that pp150 tegument protein binds to the capsid triplex with its LHB. (B) The model shows the interaction between pp150 (blue), SCP (dark blue), and MCP (brown) |
UL82是第一个以转激活因子为特征的HCMV被膜蛋白, 它直接激活早期启动子并启动病毒基因表达[59]。UL82可与视网膜母细胞瘤蛋白结合, 导致其降解, 并增强细胞周期的进展。此外, UL82还可以与细胞DAxx蛋白相互作用以促进病毒转录[60]。2017年, 舒红兵课题组[61]使用RNAi击倒策略, 发现UL82的敲除会促进宿主细胞中抗HCMV基因的转录。研究人员进一步通过内源性共免疫沉淀实验发现UL82是STING-依赖性的抗病毒反应的负调节因子。UL82可抑制cAGS (胞浆传感器)识别启动抗病毒反应, 还可与STING相互作用, 并通过两种机制损害STING介导的信号传导。① UL82通过破坏STNG-iRhom2-TRAPβ复合物抑制STING从内质网到核周微粒体的转移; ② UL82破坏了TBK1和IRF3对STING复合体的招募进而产生了免疫逃避(图 11)。该研究为抗HCMV病毒感染的疫苗设计和抗病毒药物的开发提供了新靶点, 对进一步研究具有重要意义。

|
Figure 11 The mechanism of action |
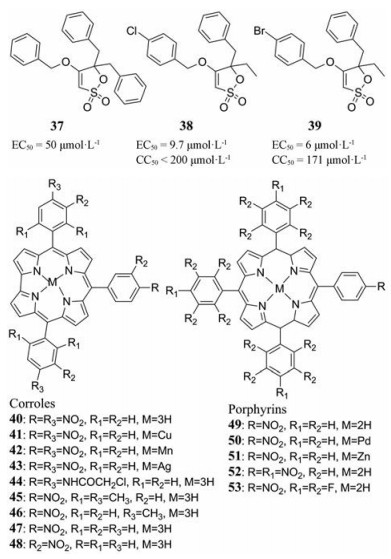
De Castro等[62]基于前期对β-氨基-γ-磺内酯化合物的研究, 发现了化合物37。37在细胞培养中对HCMV复制的抑制活性非常有限(EC50 = 50 μmol·L-1), 对很多其他DNA和RNA病毒均无活性。构效关系研究发现37磺内酯部分的C-4和C-5位置的取代基对化合物的活性及毒性的影响较大, 其中4-氯和4-溴苄氧基取代的磺内酯衍生物38和39抑制HCMV的EC50分别为9.7和6 μmol·L-1, CC50分别为 < 200和171 μmol·L-1, 是最为有效的选择性抑制剂。由于化合物38和39对多种耐药的HCMV毒株具有同样的抑制作用, 表明这些化合物可能具有新作用机制。
2015年, Gros等[63]在对咔咯(corrole)和卟啉(porphyrin)大环化合物研究的基础上, 探究了该类化合物对HCMV的抑制活性。研究人员对含有200多种卟啉类衍生物进行筛选具有抑制TB40-GFP毒株的化合物(代表化合物40~53), 结果发现40和48对HCMV病毒具有较强的抑制作用, IC50均低于0.5 μmol·L-1。此外, 这些化合物还具有较好的抗人原代成纤维细胞(MRC5)增殖活性, 在小鼠体内无明显毒性。本研究首次报道了具有抗HCMV活性的硝基咔咯类化合物, 为发现抗耐药性化合物提供了新的结构。
|
目前临床上现有的抗HCMV药物大多数作用靶点单一, 具有严重的交叉耐药性, 急需开发出高效、低毒及抗耐药性的药物及其他疗法。通过本文对抗HCMV病毒现有药物、新靶点及小分子抑制剂的研究进展的总结, 发现当前抗HCMV药物的研发“崭露头脚”。伴随着生物科学, 信息科学的突飞猛进以及对HCMV复制周期和致病机制的研究不断深入, 已有很多的作用靶点被发现, 如HCMV的表面糖蛋白GB以及控制病毒进入的五聚体等, 但还没有作用这些靶点的先导物。因此, 基于计算机辅助药物设计、人工智能等新技术及新药物设计策略以发现更多的活性化合物, 对抗HCMV首创药物的研发至关重要。
此外, 目前用于抗HCMV药物研发的策略主要有基于生物电子等排、前药修饰、高通量筛选经典方法。许多其他比较典型的药物设计的理念和方法(如骨架跃迁、分子杂合、与天然产物的优势片段结合、Protide前药技术等)也应用其中。例如, 利用前药设计策略优化上市药物更昔洛韦而获得伐昔洛韦, 药效和生物利用度显著提高。
最后, 抗病毒治疗的最大问题是易产生耐药性, 对抗耐药性问题已经刻不容缓。“鸡尾酒疗法”是一种有效的抗艾滋病疗法, 类似地, 控制HCMV感染也可将不同机制的活性药物联合使用以增加耐药性, 但任何药物的联合治疗都需要仔细匹配药代动力学特征, 以确保持续覆盖两种药物, 帮助延迟耐药性的出现。此外, 靶向靶标的保守性区域、形成主链氢键、靶向新结合位点、多靶标药物设计、共价结合或PROTAC等策略都可用于抗耐药性药物的研发, 以提高抗HCMV药物研发的成功率。
| [1] |
Bongarzone S, Nadal M, Kaczmarska Z, et al. Structure-driven discovery of α, γ-diketoacid inhibitors against UL89 herpesvirus terminase[J]. ACS Omega, 2018, 3: 8497-8505. |
| [2] |
Foglierini M, Marcandalli J, Perez L. HCMV envelope glycoprotein diversity demystified[J]. Front Microbiol, 2019, 10: 1005. |
| [3] |
Andrei G, De Clercq E, Snoeck R. Infect disord drug targets[J]. Infectious Disorders - Drug Targets, 2009, 9: 201-222. |
| [4] |
Kenneson A, Cannon MJ. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection[J]. Rev Med Virol, 2017, 17: 253. |
| [5] |
Britt WJ, Prichard MN. New therapies for human cytomegalovirus infections[J]. Antiviral Res, 2018, 159: 153-174. DOI:10.1016/j.antiviral.2018.09.003 |
| [6] |
Collins-McMillen D, Rak M, Buehler JC, et al. Alternative promoters drive human cytomegalovirus reactivation from latency[J]. Proc Natl Acad Sci U S A, 2019, 116: 17492-17497. |
| [7] |
Liu F, Zhou ZH, Arvin A, et al. Comparative Virion Structures of Human Herpesviruses[M]. Cambridge: Cambridge University Press, 2007.
|
| [8] |
Buscher N, Paulus C, Nevels M, et al. The proteome of human cytomegalovirus virions and dense bodies is conserved across different strains[J]. Med Microbiol Immunol, 2015, 204: 285-293. |
| [9] |
Kalejta RF. Tegument proteins of human cytomegalovirus[J]. Microbiol Mol Biol Rev, 2008, 72: 249-265. |
| [10] |
Choi KY, El-Hamdi NS, McGregor A, et al. Inclusion of the viral pentamer complex in a vaccine design greatly improves protection against congenital cytomegalovirus in the guinea pig model[J]. J Virol, 2019, 93: e01442-19. |
| [11] |
Chiuppesi F, Wussow F, Johnson E, et al. Vaccine-derived neutralizing antibodies to the human cytomegalovirus gH/gL pentamer potently block primary cytotrophoblast infection[J]. J Virol, 2015, 89: 11884-11898. DOI:10.1128/JVI.01701-15 |
| [12] |
Ciferri C, Chandramouli S, Donnarumma D, et al. Structural and biochemical studies of HCMV gH/gL/gO and pentamer reveal mutually exclusive cell entry complexes[J]. Proc Natl Acad Sci U S A, 2015, 112: 1767-1772. DOI:10.1073/pnas.1424818112 |
| [13] |
Neuber S, Wagner K, Goldner T, et al. Mutual interplay between the human cytomegalovirus terminase subunits pUL51, pUL56, and pUL89 promotes terminase complex formation[J]. J Virol, 2017, 91: e02384-16. |
| [14] |
Rebmann GM, Grabski R, Sanchez V, et al. Phosphorylation of golgi peripheral membrane protein grasp65 is an integral step in the formation of the human cytomegalovirus cytoplasmic assembly compartment[J]. MBio, 2016, 7: e01554-16. |
| [15] |
Yu X, Trang P, Shah S, et al. Dissecting human cytomegalovirus gene function and capsid maturation by ribozyme targeting and electron cryomicroscopy[J]. Proc Natl Acad Sci U S A, 2005, 102: 7103-7108. DOI:10.1073/pnas.0408826102 |
| [16] |
Balazs Z, Tombacz D, Szucs A, et al. Long-read sequencing of human cytomegalovirus transcriptome reveals RNA isoforms carrying distinct coding potentials[J]. Sci Rep, 2017, 7: 15989. |
| [17] |
Collins-McMillen D, Chesnokova L, Lee BJ, et al. HCMV infection and apoptosis: how do monocytes survive HCMV infection[J]. Viruses, 2018, 10: 533-551. |
| [18] |
Gentry BG, Gentry SN, Jackson TL, et al. Phosphorylation of antiviral and endogenous nucleotides to di- and triphosphates by guanosine monophosphate kinase[J]. Biochem Pharmacol, 2011, 81: 43-49. DOI:10.1016/j.bcp.2010.09.005 |
| [19] |
De Clercq E, Holy A. Acyclic nucleoside phosphonates: a key class of antiviral drugs[J]. Nat Rev Drug Discov, 2005, 4: 928-940. |
| [20] |
Fisher CE, Knudsen JL, Lease ED, et al. Risk factors and outcomes of ganciclovir-resistant cytomegalovirus infection in solid organ transplant recipients[J]. Clin Infect Dis, 2017, 65: 57-63. |
| [21] |
Manicklal S, Emery VC, Lazzarotto T, et al. The "silent" global burden of congenital cytomegalovirus[J]. Clin Microbiol Rev, 2013, 26: 86-102. |
| [22] |
Cihlar T, Chen MS. Identification of enzymes catalyzing two-step phosphorylation of cidofovir and the effect of cytomegalovirus infection on their activities in host cells[J]. Mol Pharmacol, 1996, 50: 1502-1510. |
| [23] |
Jacobson MA. Treatment of cytomegalovirus retinitis in patients with the acquired immunodeficiency syndrome[J]. N Engl J Med, 1997, 337: 105-114. |
| [24] |
Kotton CN. Updates on antiviral drugs for cytomegalovirus prevention and treatment[J]. Curr Opin Organ Transplant, 2019, 24: 469-475. DOI:10.1097/MOT.0000000000000666 |
| [25] |
Sun K, Xu H, Hilfinger JL, et al. Improved protease-targeting and biopharmaceutical properties of novel prodrugs of ganciclovir[J]. Mol Pharm, 2018, 15: 410-419. |
| [26] |
Kropeit D, Scheuenpflug J, Erb-Zohar K, et al. Pharmacokinetics and safety of letermovir, a novel anti-human cytomegalovirus drug, in patients with renal impairment[J]. Br J Clin Pharmacol, 2017, 83: 1944-1953. |
| [27] |
Geary RS, Henry SP, Grillone LR. Fomivirsen: clinical pharmacology and potential drug interactions[J]. Clin Pharmacokinet, 2002, 41: 255-260. DOI:10.2165/00003088-200241040-00002 |
| [28] |
Vanarsdall AL, Johnson DC. Human cytomegalovirus entry into cells[J]. Curr Opin Virol, 2012, 2: 37-42. |
| [29] |
Malito E, Chandramouli S, Carfi A. From recognition to execution-the HCMV pentamer from receptor binding to fusion triggering[J]. Curr Opin Virol, 2018, 31: 43-51. |
| [30] |
Stevenson EV, Collins-McMillen D, Kim JH, et al. HCMV reprogramming of infected monocyte survival and differentiation: a goldilocks phenomenon[J]. Viruses, 2014, 6: 782-807. DOI:10.3390/v6020782 |
| [31] |
Pari GS. Nuts and bolts of human cytomegalovirus lytic DNA replication[J]. Curr Top Microbiol Immunol, 2008, 325: 153-166. |
| [32] |
Sahu PK, Umme T, Yu J, et al. Selenoacyclovir and selenoganciclovir: discovery of a new template for antiviral agents[J]. J Med Chem, 2015, 58: 8734-8738. |
| [33] |
Tenney DJ, Yamanaka G, Voss SM, et al. Lobucavir is phosphorylated in human cytomegalovirus-infected and -uninfected cells and inhibits the viral DNA polymerase[J]. Antimicrob Agents Chemother, 1997, 41: 2680-2685. |
| [34] |
Zhou S, Breitenbach JM, Borysko KZ, et al. Synthesis and antiviral activity of (Z)- and (E)-2, 2-[bis(hydroxymethyl)cyclopropylidene]methylpurines and -pyrimidines: second-generation methylenecyclopropane analogues of nucleosides[J]. J Med Chem, 2004, 47: 566-575. DOI:10.1021/jm030316s |
| [35] |
Zhou S, Drach JC, Prichard MN, et al. (Z)- and (E)-2-(1, 2-Dihydroxyethyl)methylenecyclopropane analogues of 2′-deoxyadenosine and 2′-deoxyguanosine. Synthesis of all stereoisomers, absolute configuration, and antiviral activity[J]. J Med Chem, 2009, 52: 3397-3407. DOI:10.1021/jm900126v |
| [36] |
Luo M, Groaz E, Andrei G, et al. Expanding the antiviral spectrum of 3-fluoro-2-(phosphonomethoxy)propyl acyclic nucleoside phosphonates: diamyl aspartate amidate prodrugs[J]. J Med Chem, 2017, 60: 6220-6238. |
| [37] |
Luo M, Groaz E, De Jonghe S, et al. Amidate prodrugs of cyclic 9-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]adenine with potent anti-herpesvirus activity[J]. ACS Med Chem Lett, 2018, 9: 381-385. |
| [38] |
Loregian A, Appleton BA, Hogle JM, et al. Residues of human cytomegalovirus DNA polymerase catalytic subunit UL54 that are necessary and sufficient for interaction with the accessory protein UL44[J]. J Virol, 2004, 78: 158-167. |
| [39] |
Chen H, Coseno M, Ficarro SB, et al. A small covalent allosteric inhibitor of human cytomegalovirus DNA polymerase subunit interactions[J]. ACS Infect Dis, 2017, 3: 112-118. |
| [40] |
Kralj A, Wetzel A, Mahmoudian S, et al. Identification of novel allosteric modulators for the G-protein coupled US28 receptor of human cytomegalovirus[J]. Bioorg Med Chem Lett, 2011, 21: 5446-5450. |
| [41] |
Kralj A, Nguyen MT, Tschammer N, et al. Development of flavonoid-based inverse agonists of the key signaling receptor US28 of human cytomegalovirus[J]. J Med Chem, 2013, 56: 5019-5032. |
| [42] |
Ninomiya M, Tanaka K, Tsuchida Y, et al. Increased bioavailability of tricin-amino acid derivatives via a prodrug approach[J]. J Med Chem, 2011, 54: 1529-1536. |
| [43] |
Zuhlsdorf M, Hinrichs W. Assemblins as maturational proteases in herpesviruses[J]. J Gen Virol, 2017, 98: 1969-1984. |
| [44] |
Massari S, Mercorelli B, Sancineto L, et al. Design, synthesis, and evaluation of WC5 analogues as inhibitors of human cytomegalovirus immediate-early 2 protein, a promising target for anti-HCMV treatment[J]. ChemMedChem, 2013, 8: 1403-1414. |
| [45] |
Krosky PM, Borysko KZ, Nassiri MR, et al. Phosphorylation of beta-D-ribosyl benzimidazoles is not required for activity against human cytomegalovirus[J]. Antimicrob Agents Chemother, 2002, 46: 478-486. |
| [46] |
Dittmer A, Bogner E. Analysis of the quaternary structure of the putative HCMV portal protein PUL104[J]. Biochemistry, 2005, 44: 759-765. |
| [47] |
Underwood MR, Ferris RG, Selleseth DW, et al. Mechanism of action of the ribopyranoside benzimidazole GW275175X against human cytomegalovirus[J]. Antimicrob Agents Chemother, 2004, 48: 1647-1651. |
| [48] |
Reefschlaeger J, Bender W, Hallenberger S, et al. Novel non-nucleoside inhibitors of cytomegaloviruses (BAY 38-4766): in vitro and in vivo antiviral activity and mechanism of action[J]. J Antimicrob Chemother, 2001, 48: 757-767. DOI:10.1093/jac/48.6.757 |
| [49] |
Chou S. Comparison of cytomegalovirus terminase gene mutations selected after exposure to three distinct inhibitor compounds[J]. Antimicrob Agents Chemother, 2017, 61: 1-11. |
| [50] |
DFLD Silva, Cardoso JF, Silva SPD, et al. HCMV UL97 phosphotransferase gene mutations may be associated with antiviral resistance in immunocompromised patients in Belém, PA, Northern Brazil[J]. Rev Soc Bras Med Trop, 2018, 51: 141-145. |
| [51] |
Fischer L, Imrich E, Sampaio KL, et al. Identification of resistance-associated HCMV UL97- and UL54-mutations and a UL97-polymporphism with impact on phenotypic drug-resistance[J]. Antiviral Res, 2016, 131: 1-8. |
| [52] |
Sharma M, Bender BJ, Kamil JP, et al. Human cytomegalovirus UL97 phosphorylates the viral nuclear egress complex[J]. J Virol, 2015, 89: 523-534. |
| [53] |
Biron KK, Harvey RJ, Chamberlain SC, et al. Potent and selective inhibition of human cytomegalovirus replication by 1263W94, a benzimidazole l-riboside with a unique mode of action[J]. Antimicrob Agents Chemother, 2002, 46: 2365-2372. |
| [54] |
Marschall M, Stein-Gerlach M, Freitag M, et al. Direct targeting of human cytomegalovirus protein kinase pUL97 by kinase inhibitors is a novel principle of antiviral therapy[J]. J Gen Virol, 2002, 83: 1013-1023. |
| [55] |
Herget T, Freitag M, Morbitzer M, et al. Novel chemical class of pUL97 protein kinase-specific inhibitors with strong anticytomegaloviral activity[J]. Antimicrob Agents Chemother, 2004, 48: 4154-4162. DOI:10.1128/AAC.48.11.4154-4162.2004 |
| [56] |
Browne EP, Wing B, Shenk TJ, et al. Altered cellular mRNA levels in human cytomegalovirus-infected fibroblasts: viral block to the accumulation of antiviral mRNAs[J]. J Virol, 2001, 75: 12319-12330. |
| [57] |
Zhu H, Cong JP, Yu D, et al. Inhibition of cyclooxygenase 2 blocks human cytomegalovirus replication[J]. Proc Natl Acad Sci U S A, 2002, 99: 3932-3937. |
| [58] |
Dai X, Yu X, Gong H, et al. The smallest capsid protein mediates binding of the essential tegument protein pp150 to stabilize DNA-containing capsids in human cytomegalovirus[J]. PLoS Pathog, 2013, 9: e1003525. |
| [59] |
Bresnahan WA, Shenk TE. UL82 virion protein activates expression of immediate early viral genes in human cytomegalovirus-infected cells[J]. Proc Natl Acad Sci USA, 2000, 97: 14506-14511. |
| [60] |
Kalejta RF, Shenk T. Proteasome-dependent, ubiquitin-independent degradation of the Rb family of tumor suppressors by the human cytomegalovirus pp71 protein[J]. Proc Natl Acad Sci U S A, 2003, 100: 3263-3268. |
| [61] |
Fu YZ, Su S, Gao YQ, et al. Human cytomegalovirus tegument protein UL82 inhibits sting-mediated signaling to evade antiviral immunity[J]. Cell Host Microbe, 2017, 21: 231-243. |
| [62] |
De Castro S, García-Aparicio C, Andrei G, et al. 4-Benzyloxy-γ-sultone derivatives: discovery of a novel family of non-nucleoside inhibitors of human cytomegalovirus and varicella zoster virus[J]. J Med Chem, 2009, 52: 1582-1591. |
| [63] |
Gros CP, Desbois N, Michelin C, et al. Synthesis and antiviral activity evaluation of nitroporphyrins and nitrocorroles as potential agents against human cytomegalovirus infection[J]. ACS Infect Dis, 2015, 1: 350-356. |