 2020, Vol. 55
2020, Vol. 55



疱疹病毒(herpesviruses)感染是一种传播范围广、社会危害性大的疾病, 发病率高达20%[1]。现在临床上只有少数几种批准的药物可以用于疱疹病毒感染的一般治疗, 并且这些药物均为DNA聚合酶抑制剂, 结构类型单一, 且病毒对当前的疗法已产生耐药性[2, 3], 因此开发新的疱疹治疗药物成为当前亟需解决的难题。目前鉴定出的9种人类疱疹病毒均能够引起人类疾病(图 1)[4]。当人体具有正常免疫功能时, 疱疹病毒感染通常不会造成危及生命的疾病, 只会引起口腔和生殖器疱疹、水痘和带状疱疹、婴儿皮疹和传染性单核细胞增多症等, 但当人体免疫系统不成熟或受损时(如感染HIV或进行器官移植后), 疱疹病毒就可能会引起发育障碍、视力和听力丧失以及危及生命的癌症、肺炎、脑炎等疾病。
对疱疹病毒生物学机制的深入认识为人们提供了许多潜在的治疗干预途径, 尽管很多研究仍处于初始阶段, 依然有力地促进了新靶标的发现及相关抑制剂的研究。在此背景下, 笔者以PubMed和中国知网为主要查询系统, 对国内外近十年来抗疱疹病毒药物化学研究方面的新进展进行了综述。
1 疱疹病毒生物学 1.1 病毒分类所有疱疹病毒都是包膜双链DNA病毒, 病毒基因组由125~290 kbp的线性链组成, 包含约70~200个蛋白质编码基因, 其具体数量与病毒类型相关。疱疹病毒粒子(传染性颗粒)的直径约为200 nm, 主要包含3种成分:核衣壳、被膜和包膜。疱疹病毒的核衣壳(T = 16)是一个由162个多聚体(150个六聚体和12个五聚体)组成的二十面体, 内部含有病毒基因组; 被膜是一种病毒蛋白基质, 它存在于脂质双层包膜和核衣壳之间; 包膜中含有对吸附和进入细胞至关重要的糖蛋白[5]。
1.2 亚科疱疹病毒科包括感染哺乳动物、鸟类和爬行动物的疱疹病毒, 但不包括感染鱼类和青蛙的疱疹病毒(异型疱疹病毒科)或双壳类病毒(蜥蜴病毒科)。根据生物学和遗传学差异, 可以将人类疱疹病毒(human herpes virus, HHV)进一步分为3个亚科, 即α、β和γ-疱疹病毒(图 1)。迄今已鉴定出的9种人类疱疹病毒分别是:单纯疱疹病毒1和2 (HSV-1和HSV-2, α)、水痘带状疱疹病毒(VZV, α)、人巨细胞病毒(HCMV, β)、人类疱疹病毒6a、6b和7 (HHV6a、6b、7)、Epstein-Barr病毒(EBV, γ)和卡波西氏肉瘤相关疱疹病毒(KSHV, γ)。所有这些病毒都建立了终生潜伏感染, 能够周期性裂解再激活, 并且都具有致病性。这几种按亚科分类的人类疱疹病毒以及由它们引起的疾病和治疗药物如图 1所示[4]。

|
Figure 1 Human herpesviruses, diseases, and anti-herpesvirustreatments organized by subfamilies (α, β, and γ) |
α-人类疱疹病毒(HSV-1、HSV-2和VZV, 图 1)在周围神经系统的细胞中建立潜伏感染。HSV-1是口腔疱疹的主要病因, 主要存在于三叉神经节; HSV-2是生殖器疱疹的主要病原体, 更易存在于骶神经节; VZV是水痘和带状疱疹的病原体, 主要潜伏感染三叉神经节和背侧基底节。其中, HSV-1和HSV-2的原发感染、裂解复制都发生在黏液上皮细胞中。与单纯疱疹病毒不同, VZV需要通过感染的T细胞从建立感染的上呼吸道黏液上皮细胞传输到最常出现疾病的皮肤细胞。
β-疱疹病毒(HCMV、HHV6a、HHV6b和HHV7, 图 1)具有复杂的细胞嗜性, 能够感染一系列免疫细胞以及内皮细胞和成纤维细胞, 可以在体内引起包括多形核白细胞(PMNLs)、巨噬细胞和单核细胞在内的外周血单核细胞(PBMC)、内皮细胞和成纤维细胞的裂解性和潜伏性感染, 其感染机制可能与造血干细胞和CD14+单核细胞有关[6]。
γ-疱疹病毒(KSHV和EBV, 图 1)感染上皮细胞、内皮细胞和B细胞。其中EBV的裂解性和潜伏性感染都发生在B细胞中, 它可以特异性地通过其包膜糖蛋白与宿主细胞上的补体受体2 (CD21)相互作用而引起B细胞的感染, 原发性感染的确切部位和EBV从B细胞转移到上皮细胞的机制仍然有待探明, 有研究推测原发性EBV感染可能由上皮细胞介导, 还有研究推测扁桃体B细胞是原发性感染的部位, 因为B细胞比上皮细胞的感染效率更高, 也有可能两者都与原发性感染有关[7]。KSHV感染CD19+B细胞会引起多中心性Castleman病和原发性渗出性淋巴瘤, 感染内皮细胞会引起卡波西氏肉瘤。以上病毒感染部位的种类之多也再次证明了γ-疱疹病毒复杂的细胞嗜性。
1.4 病毒复制周期疱疹病毒的复制周期如图 2[4]所示。在进入过程中, 核衣壳在与宿主细胞膜融合后从包膜中释放出来, 并被内化, 然后动力蛋白(dynein)和动力蛋白激活蛋白(dynactin)组成的复合物沿着微管蛋白(tubulin)的微管(microtubules)将核衣壳转运到细胞核, 衣壳与核孔结合, 并将它们的DNA基因组释放到细胞核中。

|
Figure 2 The viral replication cycle |
病毒基因组通过复制去感染细胞, 这个过程由DNA聚合酶、螺旋酶复合物和单链DNA结合蛋白(ssDNA)组成的病毒复制体来执行。细胞RNA聚合酶II产生病毒转录本, 病毒反式激活因子作为被膜的一部分引入细胞, 启动由中间-早期启动子控制的病毒基因表达的级联。这样, 多种病毒蛋白如内切核酸酶就可以迅速影响宿主mRNA的稳定性, 导致宿主蛋白表达减少, 并对病毒转录本的翻译产生竞争优势[8], 之后主要衣壳蛋白(MCP)、组装蛋白(AP)和成熟蛋白酶(PR)在细胞质中表达并移位到细胞核, 在那里它们聚集形成未成熟的衣壳(immature capsid)。然后, 未成熟的衣壳经过病毒蛋白酶的处理逐渐成熟并形成门户顶点(portal vertex), 最终由末端酶复合物包裹病毒基因组形成成熟的衣壳[9]。成熟的核衣壳一旦形成, 就会与被膜蛋白结合, 成为被膜蛋白, 并从细胞中脱离。由于病毒衣壳太大, 不能通过核孔运输, 因此疱疹病毒通过重塑宿主细胞膜而离开细胞核, 这一输出过程的部分功能由病毒激酶(viral kinases)介导完成。在病毒衣壳通过内核膜时形成初级包膜(1° envelopment), 之后初级包膜分解病毒衣壳通过运输离开核周间隙进入细胞质并形成次级包膜(2° envelopment), 最后次级包膜从细胞质移动到细胞外, 该过程需要一系列病毒蛋白协助完成。
在潜伏期内, 病毒表型的维持与疱疹病毒的类型密切相关, 并且很大程度上取决于它们潜伏感染的细胞是否进行复制。α-疱疹病毒感染周围神经系统的细胞, 由于这些细胞通常不会复制, 所以疱疹病毒不需要进行复制或分离。β-和γ-疱疹病毒在进行复制的细胞中建立潜伏感染, 因此β-和γ-疱疹病毒就需要确保其基因组能够正确复制并分离成子细胞。KSHV中潜伏期相关核抗原(LANA)的蛋白质也已经进化到可以完成这种复制、分离的过程; EBV的Epstein-Barr病毒核抗原1 (EBNA-1)蛋白起着类似的作用[10]。同样属于β-疱疹病毒的HCMV缺乏上述LANA/EBNA-1蛋白质, 但其仍能维持终生潜伏感染, 该机制目前还没有明确的解释, Mucke等[11]提出HCMV主要即刻早期1蛋白(ME1P)以类似于LANA的方式与宿主核小体结合, 并假设ME1P对于HCMV来说起着类似于上述LANA/EBNA-1的作用, 这对研究HCMV的潜伏感染机制以及抗HCMV新靶标的发现都有十分重要的意义。
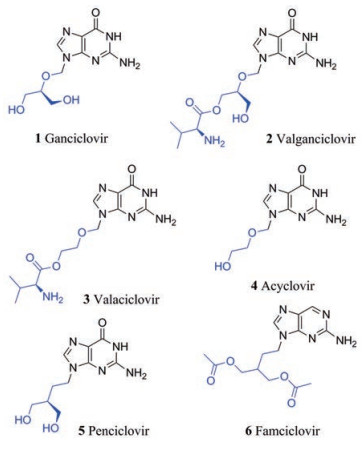
2 当前上市的抗疱疹病毒药物及作用机制目前批准的所有治疗疱疹病毒感染的方法或药物都是以病毒DNA聚合酶为靶点(除了反义硫代磷酸酯寡核苷酸福米韦生, 它仅限于眼内注射治疗HIV/AIDS患者中的HCMV视网膜炎)。更昔洛韦(ganciclovir, 1)、缬更昔洛韦(valganciclovir, 2)、伐昔洛韦(valaciclovir, 3)、阿昔洛韦(acyclovir, 4)、喷昔洛韦(penciclovir, 5)和泛昔洛韦(famciclovir, 6)都是鸟苷类似物。其中, 伐昔洛韦(3)、缬更昔洛韦(2)和泛昔洛韦(6)分别是阿昔洛韦(4)、更昔洛韦(1)和喷昔洛韦(5)的前药, 通过将缬氨酰酯或乙酸酯基团裂解来释放母体化合物[12]。不同类型的疱疹病毒有不同的病毒激酶来负责将药物磷酸化为单磷酸盐形式, 宿主激酶则进一步将单磷酸盐转化为活性的三磷酸形式, 活性三磷酸会抑制病毒DNA聚合酶的作用并结合到病毒DNA中, 从而抑制病毒DNA复制。阿昔洛韦(4)和更昔洛韦(1) (以及它们各自的前体药物)对疱疹病毒激酶的亲和力不同。例如, 更昔洛韦(1)是HCMV UL97的良好底物, 但阿昔洛韦(4)尽管仍能被UL97磷酸化, 磷酸化效果却没有更昔洛韦(1)好, 但其对于HSV胸苷激酶来说, 就是较为合适的底物了, 也正是这种可变性使得更昔洛韦(1)成为一种可供选择的抗α-疱疹病毒药物, 而阿昔洛韦(4)则通常用于治疗HCMV型疱疹病毒[13]。喷昔洛韦(5)是治疗HSV-1引起的口腔疱疹的外用药物。泛昔洛韦(6)是喷昔洛韦(5)的前药, 其口服利用度得到明显提高, 主要用于治疗带状疱疹(由VZV引起), 并在较小程度上用于复发性HSV-1和HSV-2感染。
溴夫定(brivudine, 7)、膦甲酸(foscarnet, 8)和西多福韦(cidofovir, 9)也是靶向病毒DNA聚合酶的疱疹病毒抑制剂, 但它们不属于鸟苷类似物。溴夫定(7)是一种胸苷类似物, 主要用于治疗带状疱疹, 它的功能类似于阿昔洛韦(4), 是一种三磷酸酯, 由VZV胸苷激酶单磷酸化, 随后被宿主细胞激酶转化, 它在一些欧洲和美洲国家已经获准上市[14]。西多福韦(9)是一种胞苷类似物, 与鸟苷类似物相比, 它的结构中含有单磷酸, 只需要宿主激酶直接将其磷酸化为活性二磷酸形式, 它也可以作为病毒DNA聚合酶的底物结合到病毒DNA中, 连续两个西多福韦(9)分子结合到病毒DNA中就会引起DNA链的延伸终止[15]。膦甲酸(8)是膦酸衍生物, 它不需要病毒激酶的磷酸化, 直接作用于病毒DNA聚合酶的焦磷酸结合位点, 从而防止病毒DNA链延伸, 它与西多福韦(9)都是针对抗药性疱疹病毒感染的二线治疗方法, 它们完全依赖宿主激酶将其转化为活性形式, 但是当病毒聚合酶出现耐药性突变时, 会影响这类鸟苷类似物的治疗效果。需要指出的是, 膦甲酸(8)和西多福韦(9)需要静脉给药, 并且已表现出严重的剂量限制毒性, 两者均可引起严重的肾毒性。除此之外, 西多福韦(9)还可引起骨髓抑制。因此, 新型抗疱疹病毒药物的研发依然是当前的研究热点。
|

|
疱疹病毒DNA聚合酶抑制剂是过去几十年治疗疱疹病毒感染的一线药物, 但是, 阿昔洛韦(4)的低生物利用度以及长期使用该类药物导致的耐药性问题, 使核苷类似物类抗疱疹病毒药物亟需更新换代。
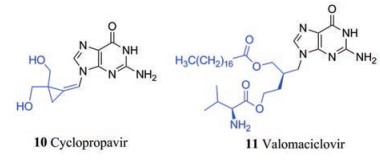
通过改进原始核苷类似物阿昔洛韦(4)得到了伐昔洛韦(3)、更昔洛韦(1)和缬更昔洛韦(2)等抗疱疹病毒药物[16]。此外, 近几年进入临床试验的鸟苷核苷类似物有cyclopropavir (10)和valomaciclovir (11)。Cyclopropavir (10)是一种二羟甲基环丙烷核苷类似物, 可与HCMV蛋白激酶UL97紧密结合并被其磷酸化, 其最终的三磷酸形式能够抑制病毒DNA聚合酶的作用从而抑制病毒复制。10具有细胞水平的抗HCMV活性及小鼠体内抗CMV的活性, 2019年7月报道的I期临床试验结果表明, 其耐受性良好, 在有效抑制浓度下无明显不良反应[17]。
|
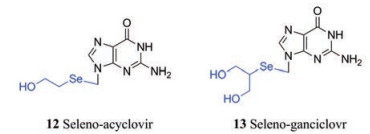
2015年, 韩国首尔大学的研究者[18]利用生物电子等排原理设计并合成了硒代阿昔洛韦(Seleno-acyclovir) (12)和硒代更昔洛韦(Seleno-ganciclovr) (13), 硒是一种对许多生物体细胞必不可少的微量元素, 它具有抗氧化性和高亲脂性, 可以增强药物的细胞渗透性, 提高口服生物利用度。体外抗病毒活性结果表明, 化合物12具有较强的抗HSV-1和HSV-2活性(EC50分别为1.47和6.34 μmol·L-1), 而化合物13具有中等的抗HCMV活性(EC50为53.1 μmol·L-1), 该研究为核苷类抗疱疹病毒药物的开发提供了新结构类型的分子, 其具体作用机制正在研究中。
|
Valomaciclovir (11)是[(R)-9[4-羟基-2-(羟甲基)丁基]-鸟嘌呤] (H2G)的前药形式, 它是20世纪90年代初发现的一种抗疱疹病毒化合物。与10不同, 11对α-疱疹病毒(HSV-1、HSV-2、VZV)和EBV均具有活性, 但对β-疱疹病毒和KSHV没有活性, 其治疗带状疱疹(VZV)和传染性单核细胞增多症(EBV)的Ⅱ期临床试验分别于2009年和2010年完成。2012年8月, 报道了其随机、双盲、活性对照试验的结果, 以已批准治疗疱疹病毒药物伐昔洛韦(3)为对照, 11在保证治疗效果的前提下能够将用药次数从每天3次减少到每天1次[19]。
最近, 俄罗斯Krasnov等[20]利用片段连接的方法设计合成了一类在Vero E6细胞中高效抑制HSV-1 (包括耐阿昔洛韦的TK株)的新化合物, 其中活性最高的是含有(R)-或(S)-7, 8-二氟-3, 4-二氢-3-甲基-2H-[1, 4]苯并噁嗪片段的嘌呤化合物(图 3), (S)-对映体(化合物14)比(R)-对映体对HSV-1表现出更高的抑制活性, (S)-对映体的EC50值为4.63 μmol·L-1, (R)-对映体的EC50值为18 μmol·L-1。

|
Figure 3 Novel purine and 2-aminopurine conjugates with chiral heterocyclic amines attached at C-6 via ω-aminoalkanoyl fragment |
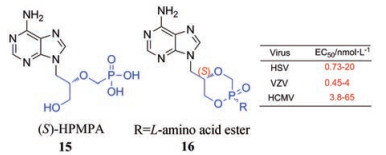
Cidofovir (9)属于无环核苷磷酸酯类似物, 本质上是一种单磷酸盐模拟物, 其注射剂主要用于治疗艾滋病患者中感染CMV导致的视网膜炎[21]。其腺嘌呤对应物, 即(S)-HPMPA (15), 是于1986年首次发现的对包括HSV、VZV在内的多种疱疹病毒具有较强抑制活性的化合物。2018年, Luo等[22]合成了一系列具有不同氨基酸基序的cHPMPA膦酰胺酸酯前体药物, 其中(S)-cHPMPA衍生的所有膦酰胺酸盐(16)都对疱疹病毒显示出广谱的抗病毒活性, 其EC50值在低纳摩尔范围内, 这些数据表明cHPMPA前体药物经过进一步开发, 有望应用于器官移植等低免疫力患者的HCMV感染治疗中。
|
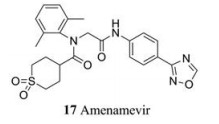
解旋酶-引物酶复合体在疱疹病毒DNA复制过程主要发挥解开病毒DNA双链以及为随后链合成引物的作用[23]。阿米那韦(amenamevir, ASP2151, 17)是一种解旋酶-引物酶抑制剂, 2011年日本富山大学Himaki课题组[24]发现ASP2151对HSV (包括对ACV耐药的HSV)和VZV感染具有疗效。2017年Kusawake等[25]发现阿米那韦的安全性和耐受性良好, 药代动力学性质优于阿昔洛韦(4), 是新一代抗疱疹病毒药物[26], 已于2017年在日本获准上市用于治疗带状疱疹。
|
终止酶将复制的病毒基因组切割成基因组长度的片段, 并将它们包装到衣壳中。它在疱疹病毒之间具有功能保守性, 且该过程在哺乳动物细胞不存在, 因此特异性的终止酶抑制剂具有毒副作用少的优点。目前终止酶抑制剂主要包括α, γ-二酮酸类衍生物和α-羟基托洛酮(α-HTs)。
3.3.1 α, γ-二酮酸类衍生物与抗HIV药物raltegravir的作用机制类似, HCMV的UL89终止酶在对衣壳内病毒DNA的加工和包装中起着关键作用, 阻断这一关键步骤可以有效抑制病毒感染, 因此, UL89终止酶可作为药物设计的新靶标。近期, 巴塞罗那大学的Bongarzone等[27]基于该靶标设计合成了一系列α, γ-二酮酸类衍生物, 代表性化合物18在低微摩尔就可以发挥体外抑制UL89终止酶活性作用(IC50 = 50 μmol·L-1), 可作为先导化合物供进一步开发, 化合物18与UL89-C结合的共晶结构如图 4所示, 18的α, γ-二酮酸片段的3个相邻的氧原子以成对的方式与UL89终止酶催化位点的两个Mn2+离子发生螯合作用, 发挥抑酶活性。

|
Figure 4 Docked conformation of 18 in the UL89-C active site |
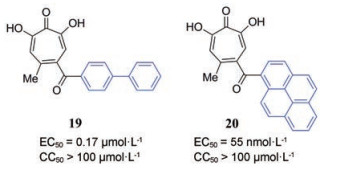
α-HTS是一种具有广泛生物活性的芳香环化合物。2018年, Dehghanpir等[28]研究了其抗疱疹病毒活性, 并推测其作用靶标可能是疱疹病毒pUL15C。2019年, 圣路易斯大学的课题组[29]合成了一系列α-HTS并研究了其初步的构效关系, α-HTS结构亲酯性有利于增加其抗病毒效力, 例如化合物19与20, 其脂水分配系数(CLogP)分别为-0.2和0.7, 相应地, 化合物20的活性显著提高。
|
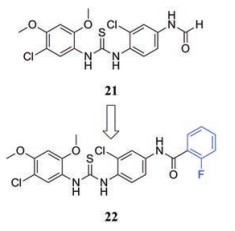
辉瑞的研究人员发现并开发了硫脲衍生物, 这类衍生物可以抑制门户顶点的形成, 经过活性筛选, 确定硫脲化合物21可以抑制HSV-1的复制, 其抑制Patton株的EC50值为7.9 μmol·L-1, 抑制E377株的EC50为24.5 μmol·L-1 [30]。对化合物21进行取代得到化合物22, 其对上述两种病毒株的效价提高了20倍[31]。这类抑制剂的作用机制尚不明确, 推测其不阻止病毒DNA复制, 而是在病毒DNA复制完成后阻止其部分基因与衣壳结合而抑制病毒的产生。
|
病毒进入宿主细胞是致病的第一步, 这一关键过程涉及到多种病毒蛋白。其中, 糖蛋白B (GB)、糖蛋白C (GC)以及硫酸乙酰肝素蛋白多糖(HSPGs)与病毒在宿主细胞表面的初始结合有关, 糖蛋白D (GD)是病毒进入宿主细胞的主要介质[32]。抑制病毒蛋白与宿主细胞的相互作用而防止病毒进入细胞已经成为在病毒感染的早期进行预防的重要方法。最近, 雅盖隆大学的研究者[33]合成了一系列不同分子量和不同取代度的阳离子葡聚糖衍生物(EXxDSy), 其中, DEX100DS40 (23)在低细胞毒性的前提下抑制HSV-1、HSV-2的EC50值分别为19.30和16.29 μg·mL-1, 能够阻止病毒粒子进入细胞从而在HSV感染早期发挥高效的抗病毒作用。
Gangji等[34]通过对HSV-1的靶向筛选从一系列称为非糖胺聚糖模拟物(NSGM)中筛选出化合物24, 其结合GD的Kd值为8~120 nmol·L-1。对HSV-1进入HeLa、HFF-1和VK2/E6E7细胞的研究结果表明该抑制剂的IC50在0.4~1.0 μmol·L-1范围内, 该研究有望为抗HSV药物的发现提供新策略。
|
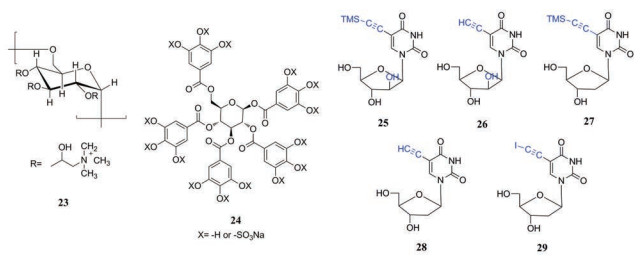
多种病毒激酶在病毒感染的整个过程均具有重要作用, 可以作为抗疱疹病毒药物的靶位。疱疹病毒的胸苷激酶(thymidine kinase, TK)参与病毒DNA的合成, 而缺失TK基因或TK基因表达能力受抑制的疱疹病毒DNA复制能力明显下降[35], 这使得TK成为抗疱疹病毒化学治疗的靶标之一。Cristofoli等[36]合成的一组阿拉伯尿苷和合成的脱氧尿嘧啶核苷的5-炔基类似物(化合物25~29), 通过抑制HSV-TK及VZV-TK而发挥抑制HSV和VZV的作用。这组化合物表现出中等的抗HSV-1活性(EC50为0.7~2 μg·mL-1), 较低的抗HSV-2活性(EC50为2~7 μg·mL-1), 同时该组化合物还表现出了明显的抗VZV活性(对OKA株的EC50为0.48~1.0 μmol·L-1)。
3.7 针对潜伏期和免疫逃避的药物目前所有应用的疱疹病毒治疗方法只针对病毒复制的过程, 而理想的治疗也会针对潜伏期。要做到这一点, 需要阻断与潜伏期维持相关的蛋白质的功能。这些蛋白质的功能可以分为两个阶段:直接维持潜伏期(例如, 允许将DNA分离成子细胞)和免疫逃避, 可作为研发抗疱疹病毒药物的重要环节[4]。维持潜伏期所需的具体蛋白与疱疹病毒的类型相关, 其中, 对EBV和KSHV的研究最深入。例如, KSHV在潜伏感染的细胞中, 仅表达对潜伏复制和持续存在所必需的一组蛋白质, 包括潜伏期相关的核抗原(LANA), 其在KSHV潜伏感染过程中发挥的作用如图 5, LANA (浅棕色)将病毒DNA (黄色和蓝色)连接到宿主组蛋白(深褐色)和附着的宿主DNA (橙色和绿色)[37]。因此, LANA也可以作为发现抗KSHV药物的靶点。

|
Figure 5 The molecular interactions between KSHV LANA, viral DNA, and host histones |
2019年, Kirsch等[37]通过使用基于片段的药物设计方法, 发现了第一个功能性LANA-DNA相互作用抑制剂, 它可以通过抑制LANA与病毒基因组之间的相互作用使KSHV的潜伏持续终止, 降低感染病毒细胞的数量。该类抑制剂的代表性化合物30与LANA可能的作用方式如图 6, 可以看到, 由于LANA二聚体的对称组装, 来自两个蛋白链(A链和B链)中的任意一个Gln1015都可以通过充当羧基的氢键供体来促进化合物30与LANA的结合。此外, Gln1073和Val1019与化合物30的吡啶基团之间存在相互作用。Kirsch等还推测Lys1069与中心三唑基团之间可能存在的阳离子-π相互作用(图中星号表示)也增加了30与LANA之间的亲和力。这是首次报道的针对LANA-DNA结合域的抑制剂, 而且也是通过化学小分子干扰核酸大分子与大分子相互作用的成功尝试, 对于研究疱疹病毒感染的调控剂具有重要意义。

|
Figure 6 3D representation of the plausible binding pose of compound 30 |
以隐蔽结合位点为靶标是利用小分子调节蛋白质功能的一种有前景的方法。美国加州大学的Acker等[38]以KSHV蛋白酶(KSHV Pr)的Trp109的旋转异构后的隐蔽结合位点为靶标, 发现了一类能够与该隐秘位点结合的变构抑制剂, 其中化合物31具有较高的细胞水平的抗病毒活性, EC50值为23.6 μmol·L-1。化合物31与KSHV二聚体蛋白酶的低温共晶体结构(PDB Code: 5UVP)如图 7所示。该研究表明疱疹病毒蛋白酶的变构靶向策略可能成为研发新一代疱疹病毒药物的有效方法, 并为以类似方式靶向其他病毒提供参考。

|
Figure 7 The cryogenic co-crystal structure of KSHV Pr dimer and compound 31 |
和抗其他病毒药物的研究类似, 目前也已经出现了一些针对宿主蛋白的抑制疱疹病毒的药物。与靶向病毒自身蛋白的药物相比, 该类药物可以有效克服治疗过程中病毒耐药性的产生。目前研究的3个主要宿主靶点分别是细胞周期蛋白依赖性激酶(CDK)、哺乳动物雷帕霉素靶蛋白(mTOR)和环氧合酶2 (COX-2)。除此之外, 还有许多其他激酶在疱疹病毒感染期间上调, 也是潜在的治疗靶点。
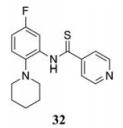
3.9.1 以CDK为靶点细胞周期蛋白依赖性激酶(CDK)是重要的细胞周期调节因子, 其中CDK1、CDK2、CDK3、CDK4和CDK6直接参与细胞周期调控, 而CDK5、CDK7、CDK8、CDK9、CDK11、CDK12和CDK13则与基因转录有关[39, 40], 当病毒感染宿主细胞时, 不同种类的CDK会对病毒进行特异性调节, 其具体作用如下: CDK1能够激活HSV-1晚期基因的表达[41]; CDK2可促进腺病毒、乳头瘤病毒和疱疹病毒DNA的复制[42]; CDK7和CDK9被证明能激活HIV和HSV的RNA转录[43-45]。因此, CDK抑制剂的研究也成为抗病毒药物开发的新方向。之前开发的CDK广谱抑制剂有Flavopiridol和Roscovitine, 然而它们因同时抑制多种CDK而引发不良反应[42]。Yamamoto等[46]研究出一种新的特异性CDK9抑制剂FIT-039 (化合物32), 它对HSV-1抑制效果显著, EC50值为0.69 μmol·L-1。Nomura等[47]对该抑制剂的安全性和有效性进行了初步评价, 试验结果表明, FIT039在有效剂量下既不影响宿主细胞的周期进程, 也不显示体内毒性。因此, FIT039是极有研究前景的抗病毒药物先导化合物。
|
mTOR是常见的抗肿瘤药物靶点, 研究发现KSHV感染宿主细胞后, 需要通过激活P13K/Akt/mTOR信号通路来维持感染[48], 这也使得mTOR成为可能的抑制KSHV靶点。中山医学院人类病毒学研究所的研究人员[49]揭示了长期使用的抗疟药物氯喹可以通过破坏mTOR和另一通路的激活而显著抑制KSHV裂解复制并诱导裂解性KSHV感染细胞的凋亡。该研究为KSHV相关疾病和KSHV与疟疾混合感染的低成本替代或联合化疗提供了有价值的信息。
3.9.3 以COX-2为靶点在KSHV、HSV、HCMV等疱疹病毒感染宿主细胞的过程中, COX-2呈现高度表达, 成为病毒存活并维持感染的重要媒介[50], 因此, 开发合适的COX-2抑制剂可用于治疗多种由疱疹病毒引起的感染, 目前COX-2抑制剂相关研究正在进行中。
4 总结与展望通过回顾近几年抗疱疹病毒药物化学的进展可以发现, 虽然我国目前仍没有新作用机制的抗疱疹病毒药物上市, 但相关研究并没有停止。目前该领域药物化学研究内容可以分为两方面:一是对已上市药物或已报道的活性化合物进行优化, 例如对经典药物阿昔洛韦的优化; 另一个方向是发现新的靶点和抑制剂, 例如通过抑制LANA与病毒基因组之间的相互作用来阻止KSHV潜伏感染的小分子抑制剂的研究。抗疱疹病毒先导化合物发现的主要途径包括筛选已有化合物、基于靶点的药物设计、基于性质的药物设计等。其中根据已知靶点设计化合物或对已确定抗疱疹病毒药物的结构修饰仍然是当前研究的一大热点, 这使得疱疹病毒DNA聚合酶抑制剂的种类与数量不断增加, 针对病毒潜伏期和针对宿主细胞的先导化合物出现使得在病毒感染早期治疗以及开发特异性抑制剂成为了可能。
在此过程中, 对抗疱疹病毒先导化合物的结构优化方法也不断“推陈出新”, 局部修饰、生物电子等排原理、对映体优化以及前药设计等方法都被应用于抗疱疹病毒药物的研究进程中, 并取得了一系列进展。
还应注意的是, 疱疹病毒的生物学复杂性不仅为抗疱疹病毒药物的发现带来了挑战, 同时也意味着许多机遇。因此, 药物的研发需要密切结合疱疹病毒生物学研究的新进展。笔者相信, 随着对病毒生物学与靶标结构生物学的深入认识以及药物设计与筛选技术的快速发展, 会出现更多、更有效的抗疱疹病毒药物。
| [1] |
Hogestyn JM, Mock DJ, Mayer-Proschel M. Contributions of neurotropic human herpesviruses herpes simplex virus 1 and human herpesvirus 6 to neurodegenerative disease pathology[J]. Neural Regen Res, 2018, 13: 211-221. |
| [2] |
Frobert E, Burrel S, Ducastelle-Lepretre S, et al. Resistance of herpes simplex viruses to acyclovir: an update from a ten-year survey in France[J]. Antiviral Res, 2014, 111: 36-41. |
| [3] |
Wilson SS, Fakioglu E, Herold BC. Novel approaches in fighting herpes simplex virus infections[J]. Expert Rev Anti Infect Ther, 2009, 7: 559-568. DOI:10.1586/eri.09.34 |
| [4] |
Gable JE, Acker TM, Craik CS. Current and potential treatments for ubiquitous but neglected herpesvirus infection[J]. Chem Rev, 2014, 114: 11382-11412. |
| [5] |
Hargett D, Shenk TE. Experimental human cytomegalovirus latency in CD14+ monocytes[J]. Proc Natl Acad Sci U S A, 2010, 107: 20039-20044. |
| [6] |
Goodrum F, Reeves M, Sinclair J, et al. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro[J]. Blood, 2007, 110: 937-945. |
| [7] |
Shu M, Taddeo B, Roizman B. The nuclear-cytoplasmic shuttling of virion host shutoff RNase is enabled by pUL47 and an embedded nuclear export signal and defines the sites of degradation of AU-rich and stable cellular mRNAs[J]. J Virol, 2013, 87: 13569-13578. DOI:10.1128/JVI.02603-13 |
| [8] |
Sattentau Q. HIV's gut feeling[J]. Nat Immunol, 2008, 9: 225-227. |
| [9] |
Newcomb WW, Homa FL, Brown JC. Involvement of the portal at an early step in herpes simplex virus capsid assembly[J]. J Virol, 2005, 79: 10540-10546. |
| [10] |
Hodin TL, Najrana T, Yates JL. Efficient replication of Epstein-Barr virus-derived plasmids requires tethering by EBNA1 to host chromosomes[J]. J Virol, 2013, 87: 13020-13028. DOI:10.1128/JVI.01606-13 |
| [11] |
Mücke K, Paulus C, Bernhardt K, et al. Human cytomegalovirus major immediate early 1 protein targets host chromosomes by docking to the acidic pocket on the nucleosome surface[J]. J Virol, 2013, 88: 1228-1248. |
| [12] |
Hagemeier SR, Dickerson SJ, Meng Q, et al. Sumoylation of the Epstein-Barr virus BZLF1 protein inhibits its transcriptional activity and is regulated by the virus-encoded protein kinase[J]. J Virol, 2010, 84: 4383-4394. |
| [13] |
Talarico CL, Burnette TC, Miller WH, et al. Acyclovir is phosphorylated by the human cytomegalovirus UL97 protein[J]. Antimicrob Agents Chemother, 1999, 43: 1941-1946. |
| [14] |
Keam SJ, Chapman TM, Figgitt DP. Brivudin (bromovinyl deoxyuridine)[J]. Drugs, 2004, 64: 2091-2099. |
| [15] |
De Clercq E. Potential of acyclic nucleoside phosphonates in the treatment of DNA virus and retrovirus infections[J]. Expert Rev Anti Infect Ther, 2003, 1: 21-43. |
| [16] |
De Clercq E. Potential antivirals and antiviral strategies against SARS coronavirus infections[J]. Expert Rev Anti Infect Ther, 2006, 4: 291-302. |
| [17] |
Rouphael NG, Hurwitz SJ, Hart M, et al. Phase 1b trial to evaluate the safety and pharmacokinetics of multiple ascending doses of filociclovir (MBX-400, cyclopropavir) in healthy volunteers[J]. Antimicrob Agents Chemother, 2019, 63: e00717-00719. |
| [18] |
Sahu PK, Umme T, Yu J, et al. Selenoacyclovir and selenoganciclovir: discovery of a new template for antiviral agents[J]. J Med Chem, 2015, 58: 8734-8738. |
| [19] |
Tyring SK, Plunkett S, Scribner AR, et al. Valomaciclovir versus valacyclovir for the treatment of acute herpes zoster in immunocompetent adults: a randomized, double-blind, active-controlled trial[J]. J Med Virol, 2012, 84: 1224-1232. |
| [20] |
Krasnov VP, Musiyak VV, Vozdvizhenskaya OA, et al. N-[ω-(Purin-6-yl)aminoalkanoyl] derivatives of chiral heterocyclic amines as promising anti-herpesvirus agents[J]. Eur J Org Chem, 2019, 30: 4811-4821. |
| [21] |
De Clercq E, Sakuma T, Baba M, et al. Antiviral activity of phosphonylmethoxyalkyl derivatives of purine and pyrimidines[J]. Antiviral Res, 1987, 8: 261-272. |
| [22] |
Luo M, Groaz E, De Jonghe S, et al. Amidate prodrugs of cyclic 9-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]adenine with potent anti-herpesvirus activity[J]. ACS Med Chem Lett, 2018, 9: 381-385. |
| [23] |
Crute JJ, Mocarski ES, Lehman IR. A DNA helicase induced by herpes simplex virus type 1[J]. Nucleic Acids Res, 1988, 16: 6585-6596. |
| [24] |
Himaki T, Masui Y, Chono K, et al. Efficacy of ASP2151, a helicase-primase inhibitor, against thymidine kinase-deficient herpes simplex virus type 2 infection in vitro and in vivo[J]. Antiviral Res, 2011, 93: 301-304. |
| [25] |
Kusawake T, Kowalski D, Takada A, et al. The influence of hepatic and renal impairment on the pharmacokinetics of a treatment for herpes zoster, amenamevir (ASP2151): Phase 1, open-label, single-dose, parallel-group studies[J]. Adv Ther, 2017, 34: 2612-2624. |
| [26] |
Yajima M, Yamada H, Takemoto M, et al. Profile of anti-herpetic action of ASP2151 (amenamevir) as a helicase-primase inhibitor[J]. Antiviral Res, 2017, 139: 95-101. |
| [27] |
Bongarzone S, Nadal M, Kaczmarska Z, et al. Structure-driven discovery of α, γ-diketoacid inhibitors against UL89 herpesvirus terminase[J]. ACS Omega, 2018, 3: 8497-8505. |
| [28] |
Dehghanpir SD, Birkenheuer CH, Yang K, et al. Broad anti-herpesviral activity of α-hydroxytropolones[J]. Vet Microbiol, 2018, 214: 125-131. |
| [29] |
Berkowitz AJ, Franson AD, GazquezCassals A, et al. Importance of lipophilicity for potent anti-herpes simplex virus-1 activity of α-hydroxytropolones[J]. MedChemComm, 2019, 10: 1173-1176. |
| [30] |
van Zeijl M, Fairhurst J, Jones TR, et al. Novel class of thiourea compounds that inhibit herpes simplex virus type 1 DNA cleavage and encapsidation: resistance maps to the UL6 gene[J]. J Virol, 2000, 74: 9054-9061. DOI:10.1128/JVI.74.19.9054-9061.2000 |
| [31] |
Newcomb WW, Brown JC. Inhibition of herpes simplex virus replication by WAY-150138: assembly of capsids depleted of the portal and terminase proteins involved in DNA encapsidation[J]. J Virol, 2002, 76: 10084-10088. |
| [32] |
Heldwein EE, Krummenacher C. Entry of herpesviruses into mammalian cells[J]. Cell Mol Life Sci, 2008, 65: 1653-1668. |
| [33] |
Pachota M, Klysik K, Synowiec A, et al. Inhibition of herpes simplex viruses by cationic dextran derivatives[J]. J Med Chem, 2017, 60: 8620-8630. |
| [34] |
Gangji RN, Sankaranarayanan NV, Elste J, et al. Inhibition of herpes simplex virus-1 entry into human cells by nonsaccharide glycosaminoglycan mimetics[J]. ACS Med Chem Lett, 2018, 9: 789-802. |
| [35] |
Zhang HG, Hanson LA. Deletion of thymidine kinase gene attenuates channel catfish herpesvirus while maintaining infectivity[J]. Virology, 1995, 209: 658-663. |
| [36] |
Cristofoli WA, Wiebe LI, De Clercq E, et al. 5-alkynyl analogs of arabinouridine and 2'-deoxyuridine: cytostatic activity against herpes simplex virus and varicella-zoster thymidine kinase gene-transfected cells[J]. J Med Chem, 2007, 50: 2851-2857. |
| [37] |
Kirsch P, Jakob V, Oberhausen K, et al. Fragment-based discovery of a qualified hit targeting the latency-associated nuclear antigen of the oncogenic Kaposi's Sarcoma-associated herpesvirus/human herpesvirus 8[J]. J Med Chem, 2019, 62: 3924-3939. |
| [38] |
Acker TM, Gable JE, Bohn MF, et al. Allosteric inhibitors, crystallography, and comparative analysis reveal network of coordinated movement across human herpesvirus proteases[J]. J Am Chem Soc, 2017, 139: 11650-11653. |
| [39] |
Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm[J]. Nat Rev Cancer, 2009, 9: 153-166. |
| [40] |
Kohoutek J, Blazek D. Cyclin K goes with CDK12 and CDK13[J]. Cell Div, 2012, 7: 12. |
| [41] |
Evers DL, Breitenbach JM, Borysko KZ, et al. Inhibition of cyclin-dependent kinase 1 by purines and pyrrolo[2, 3-d]pyrimidines does not correlate with antiviral activity[J]. Antimicrob Agents Chemother, 2002, 46: 2470-2476. |
| [42] |
Schang LM. Cyclin-dependent kinases as cellular targets for antiviral drugs[J]. J Antimicrob Chemother, 2002, 50: 779-792. |
| [43] |
Rechter S, Scott GM, Eickhoff J, et al. Cyclin-dependent kinases phosphorylate the cytomegalovirus RNA export protein pUL69 and modulate its nuclear localization and activity[J]. J Biol Chem, 2009, 284: 8605-8613. |
| [44] |
Ou M, Sandri-Goldin RM. Inhibition of CDK9 during herpes simplex virus 1 infection impedes viral transcription[J]. PLoS One, 2013, 8: e79007. |
| [45] |
Thomas JP, Tutsch KD, Cleary JF, et al. Phase I clinical and pharmacokinetic trial of the cyclin-dependent kinase inhibitor flavopiridol[J]. Cancer Chemother Pharmacol, 2002, 50: 465-472. |
| [46] |
Yamamoto M, Onogi H, Kii I, et al. CDK9 inhibitor FIT-039 prevents replication of multiple DNA viruses[J]. J Clin Invest, 2014, 124: 3479-3488. |
| [47] |
Nomura T, Sumi E, Egawa G, et al. The efficacy of a cyclin dependent kinase 9 (CDK9) inhibitor, FIT039, on verruca vulgaris: study protocol for a randomized controlled trial[J]. Trials, 2019, 20: 489. |
| [48] |
Wang L, Damania B. Kaposi's sarcoma-associated herpesvirus confers a survival advantage to endothelial cells[J]. Cancer Res, 2008, 68: 4640-4648. |
| [49] |
Yang M, Huang L, Li X, et al. Chloroquine inhibits lytic replication of Kaposi's sarcoma-associated herpesvirus by disrupting mTOR and p38-MAPK activation[J]. Antiviral Res, 2016, 133: 223-233. |
| [50] |
Sharma-Walia N, Paul AG, Bottero V, et al. Kaposi's sarcoma associated herpes virus (KSHV) induced COX-2: a key factor in latency, inflammation, angiogenesis, cell survival and invasion[J]. PLoS Pathog, 2010, 6: e1000777. DOI:10.1371/journal.ppat.1000777 |