 2020, Vol. 55
2020, Vol. 55



2. 潍坊医学院药学院, 山东 潍坊 261053
2. School of Pharmacy, Weifang Medical University, Weifang 261053, China
乙型病毒性肝炎(viral hepatitis type B), 简称乙肝(hepatitis B), 是由乙型肝炎病毒(HBV)所致的重大传染性疾病, 长期发展可导致急慢性病毒性肝炎、肝代谢失常、肝硬化和原发性肝细胞癌[1]。据世界卫生组织报道, 全球近20亿人曾感染过HBV, 其中约2.5亿人为慢性HBV感染者[2]。我国约有9 000万乙肝病毒携带者, 平均每年约28万患者因感染乙肝病毒而死亡[3]。
HBV完整的生命周期包括:吸附、穿入、脱壳、修补、转录、翻译、衣壳蛋白装配、DNA复制、获得包膜和释放等步骤形成有感染能力的完整HBV病毒颗粒(图 1)[4, 5]。理论上, 阻断病毒复制周期的任何一个环节, 都可以实现抗病毒的目的。伴随着HBV生命周期的深入研究, 越来越多针对HBV生命周期各个关键环节的靶点被相继报道。目前, 主要包括HBV吸附侵入抑制剂、靶向于共价闭合环状DNA (covalently closed circular DNA, cccDNA)抑制剂、靶向于RNA抑制剂、抑制乙肝表面抗原(hepatitis B surface antigen, HBsAg)分泌抑制剂、核衣壳蛋白抑制剂、靶向于RNase H结构域的DNA聚合酶抑制剂、病毒成熟抑制剂、宿主抗病毒靶点抑制剂和其他靶点尚不明确的HBV抑制剂[6]。本综述承接前文[7]关于HBV衣壳蛋白抑制剂的研究进展, 精选近几年具有代表性的研究实例, 从药物化学的角度总结了RNase H类及其他靶标抑制剂的前沿进展。

|
Figure 1 The life cycle of HBV |
HBV吸附和侵入抑制剂可以阻断病毒对新生肝细胞的再感染, 降低肝细胞内cccDNA的水平, 另外, 可有效地控制慢性乙肝患者肝移植后的乙肝病毒的再感染和孕妇对新生儿的传播, 有望成为根治慢性乙肝的新策略之一。
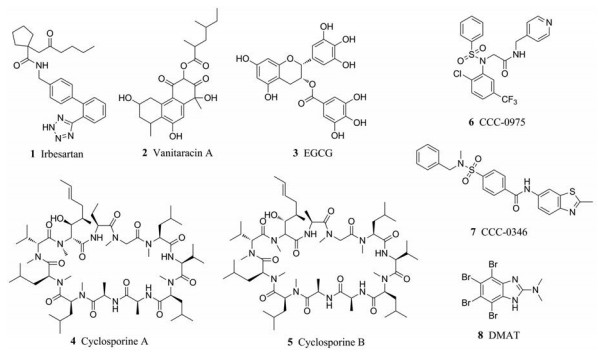
2015年, Wang等[8]研究报道, 上市药物厄贝沙坦(irbesartan, 1)通过抑制牛磺胆酸钠共转运多肽(NTCP)的表达阻断HBV进入肝细胞, 具有较好的抗病毒活性。Kaneko等[9]研究者从真菌类次生代谢化合物库中筛选得到的三环多酮类化合物2 (vanitaracin A), 具有较好的抑制HBV感染活性(EC50 = 0.61 ± 0.23 μmol·L-1, CC50 > 256 μmol·L-1)。
化合物3 (EGCG)是从绿茶中提取的一种黄酮类天然产物。研究结果表明, 其诱导NTCP受体网格蛋白依赖性内吞作用, 同时抑制转铁蛋白的内吞, 从而抑制HBV的侵入。在50 μmol·L-1浓度下, EGCG抑制HBV侵入率大于80%[10]。环孢素A (cyclosporin A, CsA) (4)及其衍生物(cyclosporin B, 5)能够抑制NTCP与大包膜蛋白结合, 阻止HBV进入肝细胞。CsA具有较好的抗HBV侵入活性, 其半数抑制浓度IC50小于0.2 μmol·L-1 [11-13]。
2 靶向于cccDNA的抑制剂cccDNA是HBV复制的起始模板, 长期存在于慢性HBV感染者肝细胞中, 也是乙肝疾病难根治和易复发的根源。清除肝细胞内cccDNA, 才能彻底抑制HBV感染, 从而实现乙肝“根治”的目标。
化合物CCC-0975 (6)和CCC-0346 (7)属于双取代磺酰胺类化合物, 均具有较好的体外抗HBV活性, 其EC50值分别为10和3 μmol·L-1。DP-rcDNA (deproteinized relaxed circular DNA)是cccDNA形成的关键中间体的前体, 也是抑制cccDNA形成的重要靶点。进一步研究结果表明, 化合物6和7可以同时减少HBV cccDNA和DP-rcDNA的水平[14]。另外, 研究发现酪蛋白激酶Ⅱ抑制剂DMAT (8)可以显著抑制细胞核的转入, 阻止cccDNA的形成[15]。
|
以RNA为靶点的药物可以有效抑制cccDNA的转录或mRNA的翻译, 是药物研究的重要靶点。小干扰RNA (siRNA)可以干扰HBV靶基因的表达和促进mRNA的降解[16, 17]。金丝桃素(helioxanthin, 9)是从贯叶连翘中提取的天然产物, 研究结果表明, 金丝桃素及其衍生物(10~12)具有较好的抗HBV活性。进一步研究结果显示, 金丝桃素可能与肝富集因子HNF 4α和HNF 3β作用, 调控前基因组RNA (pgRNA)的表达, 减少pgRNA的形成[18-20]。

|
HBsAg抑制剂包括两种, 一种是抑制蛋白的翻译, 减少HBsAg的生成; 另一种是阻止HBsAg的释放, 减少血清中HBsAg的含量[21]。其中, 文献报道较多的是抑制HBsAg的释放。
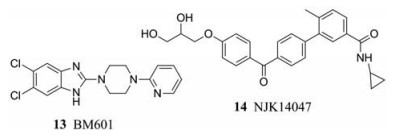
2014年, Xu等[22]通过高通量筛选得到一类苯并咪唑类化合物, 其中化合物BM601 (13)具有较好抑制HBV病毒颗粒及HBsAg分泌活性, 其EC50值分别为0.6和1.5 μmol·L-1, 但有一定的细胞毒性, 其CC50值为24.5 μmol·L-1。加拿大的REPLICor公司开发的Rep 2139和Rep 2165是核酸多聚物类HBsAg释放抑制剂, 能够显著降低HBsAg水平, 从而降低乙肝病毒载量, 目前正在Ⅱ期临床试验[23]。
2017年, Kim等[24]合成了一系列联苯酰胺类化合物, 并对其抗乙肝活性进行了评价。活性结果表明, 化合物14 (NJK14047)具有明显的体外抗HBV活性, EC50值为27 nmol·L-1, 这与HBsAg的产生、HBeAg的分泌和HBV的产生有关。NJK14047能有效抑制HBV基因转染细胞和HBV感染的人肝癌细胞中HBV抗原和HBV颗粒的分泌。与此同时, 该类化合物对HBsAg的抑制作用与p38丝裂原活化蛋白激酶(MAPK)抑制活性呈正相关。
|
HBV DNA聚合酶是一个多功能酶, 在HBV的复制周期中发挥着重要的作用, 可分为4个功能结构域:从N端到C端依次为末端蛋白区(TP)、间隔区(SD)、逆转录区(RT)和核糖核酸酶区(RNase H)[25]。其中, RT是HBV DNA聚合酶的主要功能区, 同时具有逆转录酶及DNA聚合酶活性, 可将HBV pgRNA逆转录成负链DNA, 并以此为模板合成正链DNA, 为HBV复制所必须, 是抗HBV药物开发的靶点之一。
目前研究的HBV DNA聚合酶抑制剂大多为核苷(酸)类化合物, 除了已批准上市的药物, 多个其他核苷(酸)类化合物也被广泛研究。其中核苷类化合物主要包括泛托西他滨(valtorcitabine, 15)、依夫西他滨(elvucitabine, 16)、恩曲他滨(emtrictabine, 17)和β-L-2'-F-d4C (18)等; 核苷酸类化合物主要包括开环核苷酸类似物(19)、环状核苷酸类似物(20)、核苷衍生物(21)和桥接核苷酸衍生物(22)。然而, 研究发现, 服用核苷(酸)类药物的患者需要终身用药, 而长期使用又会产生耐药性, 因此限制了核苷(酸)类抑制剂的广泛应用[26]。
RNase H结构域作为HBV DNA聚合酶的功能区之一, 在病毒复制过程中发挥着重要作用。近年来, 将RNase H结构域作为HBV治疗靶点得到了广泛关注[27, 28]。乙肝病毒在细胞质衣壳颗粒中逆转录, 病毒RNA由病毒衣壳蛋白包裹, 由病毒聚合酶催化, 复制形成负极性DNA链, 在此过程中RNase H负责降解pgRNA, 从而形成正极性DNA链。成熟的衣壳颗粒运输到细胞核形成cccDNA, 或者作为病毒粒子分泌。RNase H抑制剂会在产生负极性DNA链的过程中阻断正极性DNA链的合成, 从而抑制功能性病毒粒子的分泌和cccDNA的补充(图 2)。RNase H活性的丧失使得病毒无法正常复制, 表明HBV RNase H是潜在的药物靶标[29-31]。

|
Figure 2 Inhibition of HBV RNase H in the life cycle of HBV |
研究表明, HBV与人类免疫缺陷病毒(human immunodeficiency virus, HIV)同属于逆转录病毒, HBV和HIV RNase H同属于核苷酸转移酶家族[32]。在核心催化域内, HBV约有23%的氨基酸与HIV对应物相一致[33, 34]。Lu等[35]研究发现, 超过20%的HIV RNase H抑制剂同样对HBV RNase H有抑制活性。其中, 托酚酮类、N-羟基嘧(吡)啶二酮类和N-羟基异喹啉二酮类作为RNase H抑制剂, 均表现出潜在的抗HBV活性[36]。

|
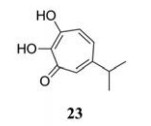
托酚酮类化合物对HIV RNase H具有较强的抑制活性[30]。β-侧柏酚(23)是一种从西部红柏树分离出的羟基化托酚酮类, 它通过螯合Mg2+离子, 与HIV-1 RNase H活性位点结合, 对HIV-1 RNase H具有抑制活性[37-39], IC50值为0.2~0.3 μmol·L-1。研究表明, β-侧柏酚对HBV也具有一定的抑制活性, 抑制D型和H型HBV RNase H活性的IC50值分别为5.9和2.3 μmol·L-1 [30]。
|
随着研究的不断深入, 羟基托酚酮逐渐发展为一类新骨架HBV RNase H抑制剂先导化合物[40]。2017年, Lu等[35]通过高通量筛选技术, 得到了一系列羟基托酚酮类衍生物, 其细胞水平的抗HBV活性均在微摩尔水平。随后, 通过进一步的研究总结出羟基托酚酮类化合物的构效关系, 左翼α-羟基酮为活性必需, 右翼R2为含羰基且为短链取代基可以增强抗乙肝活性(27, 图 3)。RNase H抑制剂可与核酸类似物起协同作用, 潜在的抗HBV RNase H药物有望与现有的核酸类似物联合使用, 抑制HBV复制, 阻止相关蛋白的表达, 从而达到根除乙肝感染的目的[41]。

|
Figure 3 The structures of hydroxytropolone derivatives 24-28 and their preliminary SAR |
二酮类HBV RNase H抑制剂包括N-羟基嘧(吡)啶二酮类(HPDs)和N-羟基异喹啉二酮类(HIDs), HPDs和HIDs作为RNase H抑制剂, 在抑制HBV活性的同时均不影响聚合酶在衣壳中延长DNA链的能力[32]。
5.2.1 N-羟基嘧(吡)啶二酮类抑制剂2017年, Edwards等[42]研究发现, HPDs可以通过靶向RNase H从而抑制HBV在细胞中的复制。几种典型的HPDs衍生物均表现出较好的抗HBV活性, 其中化合物32和33抗HBV活性最高(图 4), 在微摩尔至亚微摩尔水平发挥抑制活性, 值得进一步研究。

|
Figure 4 The structures of HPD derivatives 29-33[43] |
HIDs已被广泛应用于抗HIV RNase H研究, 并且被发现是体外抗纯化酶的有效抑制剂[44, 45]。HIDs在体内通过结合HIV RNase H活性位点上的Mg2+离子, 从而发挥抑制活性[46]。研究发现, 超过20%的HIDs化合物在体内抑制HIV RNase H活性的同时, 也具有一定的抗HBV RNase H的作用。2014年, Cai等[47]通过基于细胞活性的表型筛选得到一系列HIDs衍生物和萘啶酮类化合物(36, 图 5A), 活性结果显示, 化合物34抑制活性高, 抗HBV活性较好(EC50 = 4.2 μmol·L-1), 可作为先导化合物进行进一步的结构修饰。
2017年, Edwards等[42]通过进一步的研究, 总结出HIDs类化合物的构效关系(图 5B): ①六元含氮HPD环是最小的药效团, 替换为五元环活性大大降低。在HPD母环的5位和6位桥接一个六元环, 形成了HPD骨架。② HPD和HID基本骨架的C1~3位形成一个金属螯合位点(含氧三元组), 为活性必需。③ C4位为含有羰基的酯或酰胺基团, 通过增加相连氢的酸性, 从而增强含氧三元组螯合Mg2+的能力。与此同时, R基团为较大的疏水性基团(芳基或醛基)活性增加。

|
Figure 5 The structures of HID derivatives 34-41 and their preliminary SAR |
研究表明, HIDs骨架具有很大的发展前景, HPDs骨架也具有发展为抗HBV药物的潜力。但是, 目前已得到的抗HBV RNase H类化合物大多含有多个羟基, 极性较强, 透膜性差, 难以进入细胞发挥抗病毒作用, 从而限制了进一步的开发与应用[35]。未来的研究应在降低目标化合物细胞毒性的同时, 改善其透膜性。可以利用前药[48]的设计策略(如酯化)将其螯合基团进行封闭, 使其在细胞内相应酶解条件下释放出原药, 从而提高该类抑制剂的体内抗病毒活性。
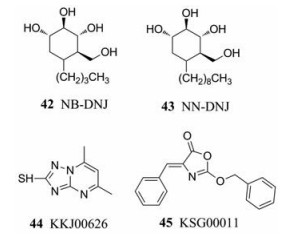
6 病毒成熟抑制剂乙肝病毒完成DNA复制的同时, 内质网核糖体产生的病毒表面蛋白在高尔基体进行修饰, 形成成熟的病毒颗粒, 最后与外膜融合将病毒释放到细胞外, 因此, 抑制乙肝病毒的成熟成为一个重要的靶点。氮端连接的多糖通过与伴侣蛋白作用促进糖蛋白的折叠, 内质网的α-葡萄糖苷酶移除葡萄糖进行糖化修饰, 因此, 抑制α-葡萄糖苷酶的活性能够阻止正常的蛋白质折叠功能。研究结果表明, NB-DNJ (42)和NN-DNJ (43, N-nonyl-DNJ)抑制α-葡萄糖苷酶, 错误折叠蛋白, 阻止蛋白的离去。通过土拔鼠乙肝动物模型的研究进一步表明, α-葡萄糖苷酶抑制剂能够干扰正常的折叠, 阻止包膜病毒颗粒的释放[49]。2016年, Asif-Ullah等[50]研究者报道了抑制病毒核心与表面蛋白作用的抑制剂, 其中化合物KKJ00626 (44)和KSG00011 (45)表现了较好的抗HBV活性。
|
乙肝病毒通过降低机体免疫应答减少病毒清除, 促使机体长期处于较弱的免疫应答, 造成肝损伤和肝功能下降, 因此, 可以通过增强宿主免疫应答和天然干扰素的产生提高抗病毒作用。目前的研究主要包括免疫激动剂、HBV治疗性疫苗、特异性CTL调节分子激动剂、热应激同源蛋白70 (Hsc70)抑制剂和靶向宿主因子抑制剂。
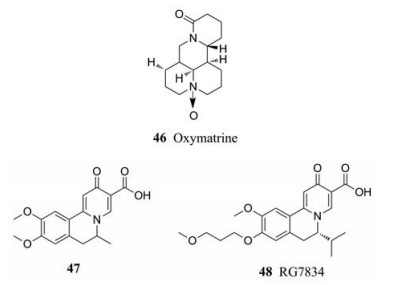
GS-9620和RO6864018属于Toll样受体7 (TLR7)激动剂, 能够持久抑制HBV抗原分泌和DNA复制, 同时增强CD8+ T细胞的杀伤活性[51]。Hsc70是一种ATP结合蛋白, 在逆转录过程中起着重要作用[52]。化合物氧化苦参碱(46, oxymatrine)是从中药苦参中提取的一种天然产物, 苦参碱及其衍生物通过破坏Hsc70 mRNA, 抑制Hsc70 mRNA的表达, 并显著抑制HBV复制。结果表明, 苦参碱对HBV野生型和耐药突变株都具有较好的抑制活性[53, 54]。
另外, 罗氏公司基于细胞水平的表型筛选, 发现二氢喹诺酮类化合物47具有较好的抗乙肝活性, EC50值为0.87 μmol·L-1。随后, 经过一系列结构修饰获得了RG7834 (48), 它通过靶向宿主因子PAPD5/7, 表现出较强的抑制HBV活性, EC50值为3.2 nmol·L-1, 目前正在Ⅰ期临
|
随着人们对抗病毒领域研究的不断深入, 一些新型的HBV抑制剂不断被开发。例如, 从抗病毒中药射干中提取分离得到的异黄酮天然产物(49)具有较好的抗HBV活性, 其抑制HBsAg分泌的EC50为53.9 μmol·L-1。2-吡喃酮衍生物50表现出较高的抑制HBeAg分泌活性(EC50 = 0.5 μmol·L-1, SI = 16.0)和较好的抑制DNA复制活性(EC50 = 2.1 μmol·L-1, SI = 3.81)。在此基础上, 通过一系列的结构优化与修饰, 构建了2-吡喃酮类活性小分子的药效团模型(图 6), 其中包括3个疏水中心(蓝色部分)、4个氢键受体(红色虚线部分)和1个氢键供体(紫色部分)[58]。

|
Figure 6 The structures of compounds 49, 50 and pharmacophore model |
目前, 已上市的抗乙肝药物多属于核苷类化合物, 随着核苷类药物在临床上的长期使用, 不断涌现出许多亟待解决的问题, 例如病毒耐药株的出现和联合用药的肾脏累积毒性, 因此, 基于新靶标新机制的抗乙肝病毒药物研发逐渐成为当前研究的热点。本综述承接前文关于HBV衣壳蛋白抑制剂研究进展, 总结了其他新靶标抑制剂研究进展, 例如HBV吸附侵入抑制剂、靶向于cccDNA抑制剂、靶向于RNA抑制剂、抑制HBsAg分泌抑制剂、靶向于RNase H结构域的DNA聚合酶抑制剂和病毒成熟抑制剂等。
利用基于优势结构的“再定位”及多样性导向合成策略获得抗HBV RNase H活性的化合物。相比于其他类型的HBV抑制剂, 靶向于RNase H类抑制剂结构简单, 易于合成, 基于先导化合物进一步优化的空间广阔, 是最有发展潜力的HBV抑制剂之一。然而目前已得到的具有抗HBV RNase H活性的化合物大多由于极性较强, 透膜性差, 无法进入细胞发挥抗病毒作用, 从而限制了其进一步的开发与应用。
近年来, 一些新的药物设计理念和技术在抗病毒领域得到了成功的应用。例如, Priscilla L. Yang课题组[59]利用靶向蛋白降解技术将特拉匹韦(telaprevir)与CRL4CRBN的配体组合形成靶向蛋白降解嵌合体(PROTAC)分子。这种新型抗病毒PROTAC分子既能抑制又能诱导丙肝病毒(HCV)的NS3/4A蛋白酶降解, 从而克服了病毒变异的问题。与此同时, 共价抑制剂作为克服病毒耐药问题的有效途径, 也逐渐受到研究者的普遍关注[60, 61]。
上述药物设计新理念为抗病毒领域的研究奠定了坚实的基础, 并对抗乙肝病毒的研究提供了重要的参考。未来应将PROTAC技术以及共价结合理念创造性地应用到抗乙肝抑制剂的研究中, 研发具有新靶标新机制的抗乙肝病毒药物。需要特别强调的是, cccDNA是HBV生命周期中稳定存在的关键因素, 只有完全清除, 才能实现乙肝的彻底治愈。因此, 运用药物筛选的新策略与新技术发现具有清除细胞核内cccDNA活性的分子, 是未来抗乙肝药物研究的重要方向, 应给予足够的重视。
| [1] |
Ott JJ, Stevens GA, Groeger J, et al. Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity[J]. Vaccine, 2012, 30: 2212-2219. DOI:10.1016/j.vaccine.2011.12.116 |
| [2] |
World Health Organization. Hepatitis B vaccines: WHO position paper, July 2017 - recommendations[J]. Vaccine, 2019, 37: 223-225. DOI:10.1016/j.vaccine.2017.07.046 |
| [3] |
Lok AS, Mcmahon BJ, Brown RS, et al. Antiviral therapy for chronic hepatitis B viral infection in adults: a systematic review and meta-analysis[J]. Hepatology, 2016, 63: 284-306. |
| [4] |
Choi IG, Yu YG. Interaction and assembly of HBV structural proteins: novel target sites of anti-HBV agents[J]. Infect Disord Drug Targets, 2007, 7: 251-256. |
| [5] |
Zoulim F. Are novel combination therapies needed for chronic hepatitis B[J]. Antiviral Res, 2012, 96: 256-259. |
| [6] |
Shih C, Chou SF, Yang CC, et al. Control and eradication strategies of hepatitis B Virus[J]. Trends Microbiol, 2016, 24: 739-749. DOI:10.1016/j.tim.2016.05.006 |
| [7] |
Ma Y, Wei FJ, Yu J, et al. Advances in research on HBV inhibitors based on new targets (1): capsid protein inhibitors[J]. Acta Pharm Sin (药学学报), 2020, 55: 554-565. |
| [8] |
Wang XJ, Hu W, Zhang TY, et al. Irbesartan, an FDA approved drug for hypertension and diabetic nephropathy, is a potent inhibitor for hepatitis B virus entry by disturbing Na+-dependent taurocholate cotransporting polypeptide activity[J]. Antiviral Res, 2015, 120: 140-146. DOI:10.1016/j.antiviral.2015.06.007 |
| [9] |
Kaneko M, Watashi K, Kamisuki S, et al. A novel tricyclic polyketide, vanitaracin A, specifically inhibits the entry of hepatitis B and D viruses by targeting sodium taurocholate cotransporting polypeptide[J]. J Virol, 2015, 89: 11945-11953. DOI:10.1128/JVI.01855-15 |
| [10] |
Huang HC, Tao MH, Hung TM, et al. (-)-Epigallocatechin-3-gallate inhibits entry of hepatitis B virus into hepatocytes[J]. Antiviral Res, 2014, 111: 100-111. DOI:10.1016/j.antiviral.2014.09.009 |
| [11] |
Watashi K, Sluder A, Daito T, et al. Cyclosporin A and its analogs inhibit hepatitis B virus entry into cultured hepatocytes through targeting a membrane transporter NTCP[J]. Hepatology, 2014, 59: 1726-1737. DOI:10.1002/hep.26982 |
| [12] |
Nkongolo S, Ni Y, Lempp FA, et al. Cyclosporin A inhibits hepatitis B and hepatitis D virus entry by cyclophilin-independent interference with the NTCP receptor[J]. J Hepatol, 2014, 60: 723-731. |
| [13] |
Iwamoto M, Watashi K, Tsukuda S, et al. Evaluation and identification of hepatitis B virus entry inhibitors using HepG2 cells overexpressing a membrane transporter NTCP[J]. Biochem Biophys Res Commun, 2014, 443: 808-813. DOI:10.1016/j.bbrc.2013.12.052 |
| [14] |
Cai D, Mills C, Yu W, et al. Identification of disubstituted sulfonamide compounds as specific inhibitors of hepatitis B virus covalently closed circular DNA formation[J]. Antimicrob Agents Chemother, 2012, 56: 4277-4288. DOI:10.1128/AAC.00473-12 |
| [15] |
Lupberger J, Schaedler S, Peiran A, et al. Identification and characterization of a novel bipartite nuclear localization signal in the hepatitis B virus polymerase[J]. World J Gastroenterol, 2013, 19: 8000-8010. DOI:10.3748/wjg.v19.i44.8000 |
| [16] |
Li GQ, Gu HX, Li D, et al. Inhibition of hepatitis B virus cccDNA replication by siRNA[J]. Biochem Biophys Res Commun, 2007, 355: 404-408. DOI:10.1016/j.bbrc.2007.01.163 |
| [17] |
Li G, Jiang G, Lu J, et al. Inhibition of hepatitis B virus cccDNA by siRNA in transgenic mice[J]. Cell Biochem Biophys, 2014, 69: 649-654. DOI:10.1007/s12013-014-9847-1 |
| [18] |
Li Y, Fu L, Yeo H, et al. Inhibition of hepatitis B virus gene expression and replication by helioxanthin and its derivative[J]. Antivir Chem Chemother, 2005, 16: 193-201. DOI:10.1177/095632020501600305 |
| [19] |
Tseng YP, Kuo YH, Hu CP, et al. The role of helioxanthin in inhibiting human hepatitis B viral replication and gene expression by interfering with the host transcriptional machinery of viral promoters[J]. Antiviral Res, 2008, 77: 206-214. |
| [20] |
Janmanchi D, Tseng YP, Wang KC, et al. Synthesis and the biological evaluation of arylnaphthalene lignans as anti-hepatitis B virus agents[J]. Bioorg Med Chem, 2010, 18: 1213-1226. DOI:10.1016/j.bmc.2009.12.038 |
| [21] |
Mohebbi A, Lorestani N, Tahamtan A, et al. An overview of hepatitis B virus surface antigen secretion inhibitors[J]. Front Microbiol, 2018, 9: 662. DOI:10.3389/fmicb.2018.00662 |
| [22] |
Xu YB, Yang L, Wang GF, et al. Benzimidazole derivative, BM601, a novel inhibitor of hepatitis B virus and HBsAg secretion[J]. Antiviral Res, 2014, 107: 6-15. DOI:10.1016/j.antiviral.2014.04.002 |
| [23] |
Al-Mahtab M, Bazinet M, Vaillant A. Safety and efficacy of nucleic acid polymers in monotherapy and combined with immunotherapy in treatment-naive bangladeshi patients with HBeAg+ chronic hepatitis B infection[J]. PLoS One, 2016, 11: e0156667. DOI:10.1371/journal.pone.0156667 |
| [24] |
Kim SY, Kim H, Kim SW, et al. An effective antiviral approach targeting hepatitis B virus with NJK14047, a novel and selective biphenyl amide p38 mitogen-activated protein kinase inhibitor[J]. Antimicrob Agents Chemother, 2017, 61: e00214-17. |
| [25] |
Lin X, Yuan ZH, Wu L, et al. A single amino acid in the reverse transcriptase domain of hepatitis B virus affects virus replication efficiency[J]. J Virol, 2001, 75: 11827-11833. |
| [26] |
Huber AD, Michailidis E, Tang J, et al. 3-Hydroxypyrimidine-2, 4-diones as novel hepatitis B virus antivirals targeting the viral ribonuclease H[J]. Antimicrob Agents Chemother, 2017, 61: e00245-17. |
| [27] |
Lomonosova E, Zlotnick A, Tavis JE. Synergistic interactions between hepatitis B virus RNase H antagonists and other inhibitors[J]. Antimicrob Agents Chemother, 2016, 61: e02441-16. |
| [28] |
Tramontano E, Corona A, Menendez-Arias L. Ribonuclease H, an unexploited target for antiviral intervention against HIV and hepatitis B virus[J]. Antiviral Res, 2019, 171: 104613. DOI:10.1016/j.antiviral.2019.104613 |
| [29] |
Tavis JE, Lomonosova E. The hepatitis B virus ribonuclease H as a drug target[J]. Antiviral Res, 2015, 118: 132-138. DOI:10.1016/j.antiviral.2015.04.002 |
| [30] |
Hu Y, Cheng X, Cao F, et al. β-Thujaplicinol inhibits hepatitis B virus replication by blocking the viral ribonuclease H activity[J]. Antiviral Res, 2013, 99: 221-229. DOI:10.1016/j.antiviral.2013.06.007 |
| [31] |
Tavis JE, Cheng X, Hu Y, et al. The hepatitis B virus ribonuclease H is sensitive to inhibitors of the human immunodeficiency virus ribonuclease H and integrase enzymes[J]. PLoS Pathog, 2013, 9: e1003125. DOI:10.1371/journal.ppat.1003125 |
| [32] |
Tavis JE, Zoidis G, Meyers MJ, et al. Chemical approaches to inhibiting the hepatitis B virus ribonuclease H[J]. ACS Infect Dis, 2019, 5: 655-658. |
| [33] |
Massari S, Corona A, Distinto S, et al. From cycloheptathiophene-3-carboxamide to oxazinone-based derivatives as allosteric HIV-1 ribonuclease H inhibitors[J]. J Enzyme Inhib Med Chem, 2019, 34: 55-74. DOI:10.1080/14756366.2018.1523901 |
| [34] |
Li MD, Bronson DL, Lemke TD, et al. Phylogenetic analyses of 55 retroelements on the basis of the nucleotide and product amino acid sequences of the pol gene[J]. Mol Biol Evol, 1995, 12: 657-670. |
| [35] |
Lu G, Lomonosova E, Cheng X, et al. Hydroxylated tropolones inhibit hepatitis B virus replication by blocking viral ribonuclease H activity[J]. Antimicrob Agents Chemother, 2015, 59: 1070-1079. DOI:10.1128/AAC.04617-14 |
| [36] |
Edwards TC, Ponzar NL, Tavis JE. Shedding light on RNase H: a promising target for hepatitis B virus (HBV)[J]. Expert Opin Ther Targets, 2019, 23: 559-563. DOI:10.1080/14728222.2019.1619697 |
| [37] |
Budihas SR, Gorshkova I, Gaidamakov S, et al. Selective inhibition of HIV-1 reverse transcriptase-associated ribonuclease H activity by hydroxylated tropolones[J]. Nucleic Acids Res, 2005, 33: 1249-1256. DOI:10.1093/nar/gki268 |
| [38] |
Beilhartz GL, Wendeler M, Baichoo N, et al. HIV-1 reverse transcriptase can simultaneously engage its DNA/RNA substrate at both DNA polymerase and RNase H active sites: implications for RNase H inhibition[J]. J Mol Biol, 2009, 388: 462-474. DOI:10.1016/j.jmb.2009.03.025 |
| [39] |
Farias RV, Vargas DA, Castillo AE, et al. Expression of an Mg2+-dependent HIV-1 RNase H construct for drug screening[J]. Antimicrob Agents Chemother, 2011, 55: 4735-4741. DOI:10.1128/AAC.00658-11 |
| [40] |
Lomonosova E, Daw J, Garimallaprabhakaran AK, et al. Efficacy and cytotoxicity in cell culture of novel α-hydroxytropolone inhibitors of Hepatitis B virus ribonuclease H[J]. Antiviral Res, 2017, 144: 164-172. DOI:10.1016/j.antiviral.2017.06.014 |
| [41] |
Chang LJ, Hirsch RC, Ganem D, et al. Effects of insertional and point mutations on the functions of the duck hepatitis B virus polymerase[J]. J Virol, 1990, 64: 5553-5558. DOI:10.1128/JVI.64.11.5553-5558.1990 |
| [42] |
Edwards TC, Lomonosova E, Patel JA, et al. Inhibition of hepatitis B virus replication by N-hydroxyisoquinolinediones and related polyoxygenated heterocycles[J]. Antiviral Res, 2017, 143: 205-217. DOI:10.1016/j.antiviral.2017.04.012 |
| [43] |
Edwards TC, Mani N, Dorsey B, et al. Inhibition of HBV replication by N-hydroxyisoquinolinedione and N-hydroxypyridinedione ribonuclease H inhibitors[J]. Antiviral Res, 2019, 164: 70-80. |
| [44] |
Billamboz M, Suchaud V, Bailly F, et al. 2-Hydroxyisoquinoline-1, 3(2H, 4H)-diones (HIDs) as human immunodeficiency virus type 1 integrase inhibitors: influence of the alkylcarboxamide substitution of position 4[J]. Eur J Med Chem, 2016, 117: 256-268. |
| [45] |
Suchaud V, Bailly F, Lion C, et al. Investigation of a novel series of 2-hydroxyisoquinoline-1, 3(2H, 4H)-diones as human immunodeficiency virus type 1 integrase inhibitors[J]. J Med Chem, 2014, 57: 4640-4660. DOI:10.1021/jm500109z |
| [46] |
Desimmie BA, Demeulemeester J, Suchaud V, et al. 2-Hydroxyisoquinoline-1, 3(2H, 4H)-diones (HIDs), novel inhibitors of HIV integrase with a high barrier to resistance[J]. ACS ChemBiol, 2013, 8: 1187-1194. |
| [47] |
Cai CW, Lomonosova E, Moran EA, et al. Hepatitis B virus replication is blocked by a 2-hydroxyisoquinoline-1, 3(2H, 4H)-dione (HID) inhibitor of the viral ribonuclease H activity[J]. Antiviral Res, 2014, 108: 48-55. DOI:10.1016/j.antiviral.2014.05.007 |
| [48] |
Perez C, Daniel KB, Cohen SM. Evaluating prodrug strategies for esterase-triggered release of alcohols[J]. ChemMedChem, 2013, 8: 1662-1667. |
| [49] |
Wen YM, Lin X, Ma ZM. Exploiting new potential targets for anti-hepatitis B virus drugs[J]. Curr Drug Targets Infect Disord, 2003, 3: 241-246. DOI:10.2174/1568005033481141 |
| [50] |
Asif-Ullah M, Choi KJ, Choi KI, et al. Identification of compounds that inhibit the interaction between core and surface protein of hepatitis B virus[J]. Antiviral Res, 2006, 70: 85-90. DOI:10.1016/j.antiviral.2006.01.003 |
| [51] |
Gane EJ, Lim YS, Gordon SC, et al. The oral toll-like receptor-7 agonist GS-9620 in patients with chronic hepatitis B virus infection[J]. J Hepatol, 2015, 63: 320-328. DOI:10.1016/j.jhep.2015.02.037 |
| [52] |
Wang YP, Liu F, He HW, et al. Heat stress cognate 70 host protein as a potential drug target against drug resistance in hepatitis B virus[J]. Antimicrob Agents Chemother, 2010, 54: 2070-2077. DOI:10.1128/AAC.01764-09 |
| [53] |
Gao LM, Han YX, Wang YP, et al. Design and synthesis of oxymatrine analogues overcoming drug resistance in hepatitis B virus through targeting host heat stress cognate 70[J]. J Med Chem, 2011, 54: 869-876. DOI:10.1021/jm101325h |
| [54] |
Du NN, Li X, Wang YP, et al. Synthesis, structure-activity relationship and biological evaluation of novel N-substituted matrinic acid derivatives as host heat-stress cognate 70 (Hsc70) down-regulators[J]. Bioorg Med Chem Lett, 2011, 21: 4732-4735. DOI:10.1016/j.bmcl.2011.06.071 |
| [55] |
Han X, Zhou C, Jiang M, et al. Discovery of RG7834: the first-in-class selective and orally available small molecule hepatitis B virus expression inhibitor with novel mechanism of action[J]. J Med Chem, 2018, 61: 10619-10634. DOI:10.1021/acs.jmedchem.8b01245 |
| [56] |
Mueller H, Wildum S, Luangsay S, et al. A novel orally available small molecule that inhibits hepatitis B virus expression[J]. J Hepatol, 2018, 68: 412-420. |
| [57] |
Mueller H, Lopez A, Tropberger P, et al. PAPD5/7 are host factors that are required for hepatitis B virus RNA stabilization[J]. Hepatology, 2019, 69: 1398-1411. |
| [58] |
Lv ZL, Sheng CQ, Wang TT, et al. Design, synthesis, and antihepatitis B virus activities of novel 2-pyridone derivatives[J]. J Med Chem, 2010, 53: 660-668. |
| [59] |
Wispelaere M, Du G, Donovan KA, et al. Small molecule degraders of the hepatitis C virus protease reduce susceptibility to resistance mutations[J]. Nat Commun, 2019, 10: 3468. DOI:10.1038/s41467-019-11429-w |
| [60] |
Chan AH, Lee WG, Spasov KA, et al. Covalent inhibitors for eradication of drug-resistant HIV-1 reverse transcriptase: from design to protein crystallography[J]. Proc Natl Acad Sci U S A, 2017, 114: 9725-9730. DOI:10.1073/pnas.1711463114 |
| [61] |
Windsor IW, Palte MJ, Lukesh JC, et al. Sub-picomolar inhibition of HIV-1 protease with a boronic acid[J]. J Am Chem Soc, 2018, 140: 14015-14018. DOI:10.1021/jacs.8b07366 |