 2020, Vol. 55
2020, Vol. 55



2. 潍坊医学院药学院, 山东 潍坊 261053
2. School of Pharmacy, Weifang Medical University, Weifang 261053, China
乙型肝炎病毒(hepatitis B virus, HBV)是一种嗜肝DNA病毒, 长期感染可引发慢性乙型肝炎(chronic hepatitis B, CHB)、肝代谢失常、肝硬化(liver cirrhosis, LC)和肝癌(hepatocellular carcinoma, HCC)等疾病。根据世界卫生组织(World Health Organization, WHO)的相关统计, 全球大概有2.6亿慢性乙肝患者[1], 主要分布在亚洲和非洲等地区, 我国是乙肝高发国, 慢性乙肝患者约两千万人。然而, 目前乙肝病毒仍然不能被已上市的核苷(酸)类抗HBV小分子药物完全清除。因此, 研发安全高效的非核苷类HBV抑制剂具有重要意义。
随着结构生物学的发展, HBV生命周期中多个靶点近年来被相继报道。其中, HBV衣壳蛋白在生命周期的多个关键步骤中发挥重要作用, 包括前基因组RNA (pregenomic RNA, pgRNA)逆转录、自由环状DNA (relaxed circular DNA, rcDNA)的合成和成熟病毒颗粒的形成过程。HBV衣壳蛋白由90个或120个核心二聚体以疏水作用聚集形成, 但相互作用力较弱(-2.9~-4.4 kcal·mol-1), 可作为一个药物靶点[2, 3]。衣壳蛋白抑制剂与已上市的核苷类HBV抑制剂有着不同的作用机制, 对于一些核苷类药物耐药株仍具有较好的活性, 可以与核苷类药物联用发挥协同抗病毒作用, 具有良好的发展前景, 是目前研究最多的非核苷类小分子抑制剂[4, 5]。
衣壳蛋白抑制剂又称核心蛋白变构调节剂(core protein allosteric modulators, CpAM), 其作用机制主要分为3类, 包括:干扰核衣壳的正常装配, 促进核心蛋白的降解, 如二氢嘧啶类抑制剂(heteroaryldihydropyrimidines, HAPs); 促进不含pgRNA的“空衣壳”的形成, 如苯磺酰胺类抑制剂; 结合到已装配好的衣壳蛋白上, 破坏衣壳的结构, 抑制其成熟过程以及释放HBV遗传物质(HAP-TAMRA)等。
根据不同的结构类型可分为丙烯酰胺类抑制剂、二氢嘧啶类抑制剂(HAPs)、苯磺酰胺类抑制剂(SBAs)、双噻唑类抑制剂、二甲酰噻吩类抑制剂、哒嗪酮类抑制剂、二磺酸联萘类抑制剂和亚氨基糖类抑制剂等[4-7]。
1 丙烯酰胺类抑制剂1998年, King等[8]首次报道了一种新型、高效且具有高选择性的丙烯酰胺类HBV抑制剂AT-61 (1), 在4种不同的细胞系(HepAD38、HepG2.2.15、HepG2和M539V细胞系)中均表现了较好的体外抗病毒活性, EC50值分别为1.9、5.7、2.9和0.6 μmol·L-1, 而对土拨鼠肝炎病毒(WHV)、鸭乙肝病毒(DHBV)等其他病毒均无抑制作用(EC50 > 81 μmol·L-1)。AT-61对HBV聚合酶没有抑制活性(EC50 > 81 μmol·L-1), 表明其作用机制不同于已上市的核苷类药物。
2000年, Perni等[9]以AT-61为先导化合物, 对其A环和B环进行多样性的结构修饰, 设计合成了一系列丙烯酰胺类HBV抑制剂, 其中化合物AT-130 (2)体外抗病毒活性最好(EC50 = 0.13 μmol·L-1, EC90 = 0.92 μmol·L-1, CC50 > 61 μmol·L-1), 超出AT-61 (EC50 = 1.2 μmol·L-1, EC90 = 13 μmol·L-1, CC50 > 81 μmol·L-1)十倍之多。构效关系表明, A环邻位取代活性高于对位取代, 取代基以甲氧基活性最好; B环对位取代活性最好, 取代基以硝基最佳; 取代酰胺基团中以哌啶的活性最好; 溴代乙烯基的活性高于氯代。
2010年, Dong等[10]基于生物电子等排原理设计合成了一系列噻二唑丙烯酰胺类化合物, 其中化合物3 (EC50 = 3.59 μg·mL-1)和4 (EC50 = 9.0 μg·mL-1)的体外抑制HBV DNA复制活性较好。但是它们的细胞毒性较大, 选择系数(selection index, SI)分别为4.96和21.38。
同年, Katen等[4]报道了丙烯酰胺类小分子的作用机制, 以1和2为例, 它们可以捕获衣壳蛋白装配过程中的中间体, 并加速衣壳蛋白的正常装配过程, 最终形成不含有pgRNA和病毒聚合酶的“空衣壳”。
2011年, Wang等[11]合成了一系列丙烯酰胺衍生物, 并分离得到E构型和Z构型异构体, 绝对构型通过X单晶衍射确定。活性结果显示, Z构型化合物高于E构型化合物。化合物5的活性最好(EC90 = 5.51 μmol·L-1, CC90 > 10 μmol·L-1)。然而, 为保持优势构象, 采用成环策略设计合成的化合物6 (EC90 > 10 μmol·L-1, CC90 > 10 μmol·L-1)并未表现明显抗HBV活性。化合物AT-130活性最好, 对拉米夫定耐药株也有较好的抑制活性。

|
2013年, Katen等[12]首次报道了AT-130与HBV衣壳蛋白的晶体复合物, 这有助于基于结构的合理药物设计, 发现活性更好的丙烯酰胺类抑制剂。研究表明, AT-130能够改变蛋白的空间构象, 影响衣壳蛋白的正常装配, 形成大量的不含pgRNA的空衣壳[4, 8, 13]。从AT-130与HBV衣壳蛋白的晶体复合物可以看出, AT-130进入氨基酸残基Trp102、Tyr118和Pro25形成的疏水口袋中, 两个羰基与氨基酸残基Trp102和Ser106形成氢键作用, 另外, 硝基与Asp29和Thr33也形成氢键作用(图 1)[14]。对配体与蛋白精准结合模式的认识, 有助于对先导化合物进行合理结构优化。

|
Figure 1 Crystal complex of AT-130 and HBV capsid protein (PDB: 4G93) |
2001年, Bayer公司首次报道了二氢嘧啶类化合物Bay 41-4109 (7)具有体外抗HBV活性(HepG2.2.15细胞系中, EC50 = 0.05 μmol·L-1, CC50 = 7 μmol·L-1); 进一步研究发现Bay 41-4109在乙肝病毒转染的小鼠模型中表现出较好的体内抗病毒活性, 与拉米夫定相比可以同等程度地减少小鼠肝脏和血清中病毒DNA的含量, 且明显降低细胞质中HBcAg的含量, 表明Bay 41-4109与核苷类药物作用机制不同; Bay 41-4109在小鼠体内的口服生物利用度为30%, 在兔和犬体内的口服生物利用度约为60%;但是Bay 41-4109在兔体内表现出一定的肝毒性[15]。
2003年, Deres等[16]在Science上又报道了二氢嘧啶类似物BAY 38-7690 (8, EC50 = 0.15 μmol·L-1, CC50 = 50 μmol·L-1)和BAY 39-5493 (9, EC50 = 0.03 μmol·L-1, CC50 = 25 μmol·L-1)具有较好的抗HBV活性, 并且能干扰核心蛋白的正常装配, 促进细胞内核心蛋白的降解。
2005年, Stray等[17]在PNAS上报道了一个代表性的二氢嘧啶类似物HAP-1, 其可通过变构调节作用导致核衣壳的异常装配; 2006年, Bourne等[18]报道了HAP-1 (10)与HBV衣壳蛋白的晶体复合物(PDB代码: 2G34), 进一步阐述了HAP-1的作用机制, 其在衣壳蛋白亚基之间的界面以疏水作用力相结合, 轻微的改变接触面的几何形状, 影响HBV核衣壳的组装。
2008年, Bourne等[19]设计合成了一系列结构相关的化合物, 并评价了抑制HBV DNA复制活性, 其中HAP-12 (11)的活性最好, EC50值为0.05 ± 0.01 μmol·L-1 (HepG2.2.15细胞系)。
2009年, Zhu等[20]基于已报道的HAP类抑制剂, 采用并环的策略设计合成了一批结构类似物, 其中化合物12活性较好(EC50 = 0.44 μmol·L-1, CC50 = 623.80 μmol·L-1)。虽然已有的HAP类化合物具有较好的体内外抗病毒活性, 但是其水溶性较差, 影响了其药代动力学性质。为了在保持HAP抗病毒活性的同时增加其水溶性, 李松等[21]在二氢嘧啶骨架上引入一些助溶性的基团, 在所得到的化合物中, 13的活性最好(EC50 = 2.35 μmol·L-1, CC50 = 31.48 μmol·L-1), 并可诱导衣壳蛋白的异常装配。此外, 文献还报道二氢嘧啶环5位以碘原子取代的HAP衍生物也表现出一定的细胞活性(14, HepAD38细胞系, EC50 = 4.0 μmol·L-1, HepG2细胞系, CC50 > 100 μmol·L-1)[22]。
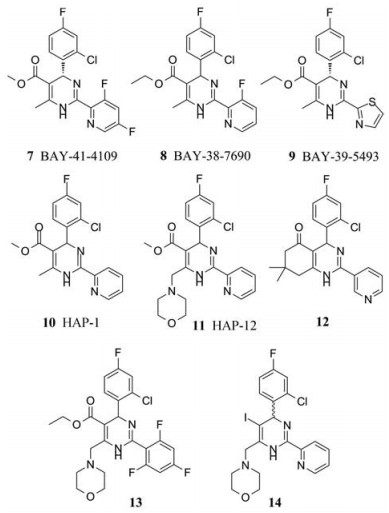
|
第一代二氢嘧啶类化合物的构效关系研究表明, 4位的2′-溴-4′-氟取代活性最好, 且4′-氟取代是活性必需; 1位NH是活性必须; 2位脂肪烃取代活性消失, 噻唑活性最好; 酯基的修饰会导致活性降低, 甲基或乙基活性最好; S构型较R构型活性好。取代基微小的改变可能对活性影响较大, 可能与蛋白-蛋白相互作用有关。此外, Tu等[23]还建立出二氢嘧啶类化合物的三维定量结构活性关系模型(3D quantitative structure activity relationship, 3D-QSAR)。
在探索二氢嘧啶类化合物构效关系的同时, 机制研究表明, 二氢嘧啶类抑制剂作为衣壳蛋白装配调节剂[24], 通过诱导形成不含pgRNA的空衣壳, 并破坏正常衣壳蛋白的稳定性抑制HBV的复制[21]。化合物HAP1能够导致衣壳蛋白四级结构的改变, 活化衣壳蛋白的装配和错误组装[25]。
总体来看, 第一代二氢嘧啶类衣壳蛋白抑制剂毒性较强, 理化性质差, 但同时抗病毒活性水平达到了纳摩尔级别, 值得进一步研究。随着研究的深入, 以二氢嘧啶环2位噻唑取代、6位吗啉取代为代表的活性更好的第2代二氢嘧啶类小分子陆续被发现。
2.2 第2代二氢嘧啶类化合物2012年, Wu等[26]报道了化合物GLS4 (15, HepAD38细胞系中, EC50 = 62.24 nmol·L-1; 原代肝细胞, CC50 = 115 μmol·L-1), 其抑制HBV DNA复制活性和细胞毒性优于Bay 41-4109 (HepAD38细胞系中, EC50 = 124.28 nmol·L-1; 原代肝细胞, CC50 = 35 μmol·L-1); 进一步研究表明GLS4抑制细胞内HBcAg含量的活性高于Bay 41-4109;但体内药代动力学性质较差, 以10 mg·kg-1的剂量对小鼠口服给药时, 半衰期为1.78 h, 生物利用度为25.5%, GLS4已进入Ⅱ期临床研究。
2015年, Klumpp等[27]报道了NVR-010-001-E2 (16), 抗病毒活性高于GLS4, EC50值为11 nmol·L-1 (HepG2.2.15细胞系), 同时对一些耐药株也都具有较好的抗HBV活性; NVR-010-001-E2与核心蛋白的结合模式也被解析出来。从共晶结构(图 2)可以看出: NVR-010-001-E2的二氢嘧啶母环通过3位氮原子与Trp102形成氢键, 二氢嘧啶环上的各个取代基与蛋白之间还存在额外作用力; 4位的2′-氟-4′-溴取代的苯基位于由Pro25、Asp29、Leu30、Thr33、Trp102、Ile105和Ser106残基形成的较大的疏水口袋(橙色), 溴原子指向口袋的深处, 氟原子与相邻亚基的Val124残基相互作用; 2位的噻唑环伸入由Phe23、Trp102、Tyr118和Phe122等残基构成的疏水腔内(红色), 氮原子与Tyr118形成氢键, 同时噻唑基团与相邻亚基的Thr128也存在着作用力; 5位的酯基位于Thr109、Phe110、Thr33和Leu37形成的腔穴顶部(青色), 而单体氧原子暴露于溶剂中; 吗啉环与相邻亚基的Val124、Trp125、Thr128和Pro134相互作用。

|

|
Figure 2 Binding model of ligand NVR-010-001-E2 to capsid protein (PDB:5E0I) |
与第1代相比, 第2代二氢嘧啶类抑制剂的抗病毒活性进一步提高, 其骨架中的噻唑与吗啉环均能与周围的氨基酸残基形成额外的作用力, 提高分子对靶标的亲和力; 同时毒性大幅度降低。但是2代HAPs的吗啉环在体内稳定性较差, 容易被氧化为无活性的嘧啶类代谢产物[28]。GLS4目前仍然停留在II期临床研究阶段, 发现体内药代性质更好的结构类似物是该领域研究的一个重要突破口。
2.3 第3代二氢嘧啶类化合物2016年, Qiu等[29]基于系统的构效关系研究发现了一系列4-甲基二氢嘧啶类化合物, 其中化合物17的HBV DNA复制抑制活性最好(EC50 = 84 nmol·L-1, CC50 = 160 μmol·L-1, HepDE19细胞系); 药代动力学研究表明17在CD-1小鼠体内的口服半衰期为0.8 h, 生物利用度为36%;进一步的结构生物学研究阐明了17与HBV衣壳蛋白的结合模式(PDB代码: 5GMZ), 如图 3, 二氢嘧啶6位的四氢吡咯环朝向溶剂开口区, 环上的羧基与Ser141形成氢键作用。2017年, 该课题组又继续报道了溶剂开口区含羧基取代的二氢嘧啶类衍生物, 化合物18 (EC50 = 3 nmol·L-1, CC50 > 100 μmol·L-1)和19 (EC50 = 3 nmol·L-1, CC50 = 65 μmol·L-1)体外活性最好, 且水溶性较好, LogP值分别为0.3和1.4。18在小鼠体内的药代动力学性质较好, 以3 mg·kg-1的剂量口服给药时, 半衰期为1.5 h, 口服生物利用度为37%;同样以3 mg·kg-1的剂量对水动力注射(hydrodynamic injection, HDI)模型小鼠口服给药, 经过5天的培养, 18可以将血浆中的HBV DNA的含量降低100倍, 与恩替卡韦的效果相当; 18的体外安全性较好, 未表现出hERG毒性、致突变性以及其他毒性[30]。化合物19与衣壳蛋白的结合模式与17类似, 如图 3, 溶剂开口区的羧基同样可与Ser141主链形成氢键作用力(PDB代码: 5WRE)。

|
Figure 3 Crystal complex of 17 (yellow)/19 (magenta) and HBV capsid protein (PDB: 5GMZ/5WRE) |
2017年, Li等[31]报道了一系列四氢吡咯并[1, 2-c]嘧啶的稠环类HAP衍生物, 化合物20活性最好(HepG2.2.15细胞系, EC50 = 10 nmol·L-1); 其化学结构中含有一个伸向蛋白-溶剂界面的磺酰胺基团, 分子模拟显示磺酰胺基团上的一个氧原子与Ser141主链可形成氢键作用; 20在小鼠体内的药代动力学性质较好, 以10 mg·kg-1的剂量口服给药时, 生物利用度达54%, 远远高于GLS4 (F = 14%)。
2017年, 本课题组基于已报道HAP类抑制剂的构效关系和核心蛋白与配体的晶体复合物结构, 设计合成了一类HAP-三氮唑类衍生物。其中, 化合物21抑制HBV DNA复制的活性(EC50 = 0.35 ± 0.04 μmol·L-1)超过了拉米夫定(EC50 = 0.54 ± 0.18 μmol·L-1), 且细胞毒性小(CC50 > 50 μmol·L-1), 但是没有表现出对s抗原和e抗原分泌的抑制活性(IC50 > 50 μmol·L-1)[32]。
2018年, 东阳光药业报道了一个抗病毒活性较好(EC50 = 39 nmol·L-1)、安全性高(hERG, IC50 > 30 μmol·L-1)的HAP类似物22; 在大鼠体内的口服生物利用度为47.7%, 在犬体内的口服生物利用度为114.1%, 远远高于GLS4 (F = 23.8%)[33]。此外相关文献还报道了溶剂开口区为桥环的衍生物23 (EC50 = 4 nmol·L-1)和24 (EC50 = 0.7 nmol·L-1)也具有较好的活性, 有进一步开发的前景[34]。
相比第2代抑制剂, 第3代抑制剂的结构中在溶剂开口区引入了羧基等极性基团, 抗病毒活性及水溶性显著提高, 安全性和药代动力学性质较好, 部分化合物正处于临床前研究。
2.4 第4代二氢嘧啶类化合物多个HAP小分子与HBV衣壳蛋白的共晶结构显示其2位的噻唑、4位的苯基以及5位的酯基均伸入一个较大的疏水口袋, 与靶标之间形成了较强的范德华力, 这些位点的可修饰性较小, 而6位的取代基朝向广阔的溶剂开口区, 空间较大, 可修饰性较强[29], 近两年报道的新型第4代HAP小分子的结构修饰主要集中在该位点。例如, 2016年, 专利公开了一类非芳香稠环取代的HAP衍生物, 其中25~28的活性较好, EC50值分别为4、0.8、1和1 nmol·L-1 [35]; 2019年最新公开的一项专利报道了在非芳香稠环基团上连有大体积取代基的HAP衍生物, 化合物29 (EC50 = 1 nmol·L-1, CC50 > 150 μmol·L-1)抗病毒活性较好且细胞毒性较小[36]。
在结构修饰的同时, HAPs精确的作用机制也日益明确。2018年, Schlicksup等[7]将一个荧光分子拼接在HAP的溶剂开口区得到了一个新的化合物HAP-TAMRA (30), 显微镜下观察其可以结合到已装配好的衣壳蛋白上, 破坏核衣壳外层的结构, 阻止其与宿主细胞中的细胞器相互作用以及释放HBV遗传物质。同年, 美国化学会刊(JACS)也报道了一个结合了荧光分子的HAP类似物(HAP-ALEX, 31), 机制研究表明HAP可以诱导核心蛋白的错误装配, 形成较大的不规则聚合物, 改变核心蛋白在细胞中的正常分布[6]。
3 苯磺酰胺类抑制剂2013年, Campagna等[37]对含有26 900个小分子的化合物库进行高通量筛选, 发现了一系列可显著降低胞浆中HBV DNA含量的苯磺酰胺(sulfamoylbenzamide, SBA)衍生物。进一步构效关系研究表明, 多氟取代的衍生物抗HBV活性明显提高, 其中化合物DVR-23 (32, AML12HBV10细胞系, EC50 = 0.3 ± 0.05 μmol·L-1, CC50 > 50 μmol·L-1; HepDES19细胞系, EC50 = 0.1 ± 0.01 μmol·L-1, CC50 > 50 μmol·L-1)和DVR-56 (33, AML12HBV10细胞系, EC50 = 0.39 ± 0.03 μmol·L-1, CC50 > 50 μmol·L-1; HepDES19细胞系, EC50 = 0.14 ± 0.09 μmol·L-1, CC50 > 50 μmol·L-1)的活性最好。机制研究表明该类化合物可以干预pgRNA和衣壳蛋白的装配过程。另外该类化合物选择性较好, 对鸭乙型肝炎病毒(DHBV)和土拨鼠肝炎病毒(WHV)均没有抑制作用。
2017年, Sari等[38]以DVR-23为先导化合物, 在苯环和磺酰胺等多位点进行广泛的修饰, 得到27个目标化合物, 其中化合物34在HepAD38细胞系中抑制HBV DNA复制活性较好, 其EC50值为0.8 ± 0.2 μmol·L-1, EC90值为7.8 ± 0.6 μmol·L-1, HepG2细胞系CC50 = 37.1 ± 0.04 μmol·L-1; 34对e抗原的分泌、细胞内和细胞外DNA也有一定的抑制作用, 其IC50值分别为1.3 ± 0.4、0.8 ± 0.2和0.6 ± 0.002 μmol·L-1, IC90值分别为4.6 ± 2.0、7.8 ± 0.6和4.3 ± 0.07 μmol·L-1。
2018年, Wu等[39]报道一个苯磺酰胺类似物ENAN-34017 (35, WT: EC50 = 0.06 ± 0.01 μmol·L-1; F97L: EC50 = 0.05 ± 0.01 μmol·L-1; AML12HBV10细胞系: CC50 > 50 μmol·L-1), 其对野生株和一些突变株的抑制活性超过了二氢嘧啶类衣壳蛋白抑制剂Bay 41-4109 (WT: EC50 = 0.09 ± 0.01 μmol·L-1; F97L: EC50 = 0.13 ± 0.01 μmol·L-1)。同年, Vandyck等[40]报道了一个对多种HBV细胞系均有抑制作用且毒性较小的SBA衍生物36 (HepG2.2.15细胞系, EC50 = 0.12 μmol·L-1; HepG2.117细胞系, EC50 = 0.43 μmol·L-1; HepG2细胞系, CC50 > 100 μmol·L-1)。进而, 小鼠体内药代动力学实验表明, 当以50 mg·kg-1的剂量口服给药时, 36的半衰期为0.7 h, 口服生物利用度可达66%。上海Longwood生物制药公司2018年公开的一篇专利(WO2018202155)中报道了一类新型双环化合物, 可作为HBV衣壳装配抑制剂。其中化合物37抑制HBV核衣壳装配活性较高, 在HepG2.2.15细胞系中, 该化合物可显著降低细胞内HBV DNA的含量且细胞毒性较小(EC50 = 0.1~100 nmol·L-1, CC50 > 30 000 nmol·L-1)[41]。

|

|
近期, 强生公司收购了诺华开发的候选化合物NVR-3-778 (38), 正在进行Ⅱ期临床试验。NVR-3-778在HepG2.2.15细胞系中抑制HBV DNA的EC50值为0.40 ± 0.13 μmol·L-1, CC50值为14.5 ± 5.7 μmol·L-1; 对不同的耐药株也表现出较好的活性, 其中在rtL180M/M204V、rtL180M/M204V/N236T、rtA181V、rtN236T和rtA181V/N236T测得的IC50值分别为1.3 ± 0.6、1.4 ± 0.5、0.82 ± 0.19、0.85 ± 0.40和0.85 ± 0.26 μmol·L-1; 此外NVR-3-778与核苷类药物联用具有协同抗HBV效果; NVR-3-778在体外动物实验中药代动力学性质较好, 口服生物利用度达84.6%[42]。SBA结构类似物JNJ-6379也进入了Ⅱ期临床研究, 其结构与相关临床实验数据目前尚未公开[43, 44]。
二氢嘧啶类与苯磺酰胺类化合物与衣壳蛋白的作用机制并不相同, 但两种参考化合物19和38却在核心蛋白Y132A六聚体的晶体结构中的二聚体-二聚体界面处与同一口袋结合, 如图 3和4。两者结合模式的显著差异在于Phe23、Tyr118和Phe122等残基构成的疏水亚口袋, 该亚口袋被19的噻唑基占据, 但对于38来说, 这个位置并没有相似的相互作用, 这种差异提示两类化合物可能有独特的抗耐药性[5]。

|
Figure 4 Crystal complex of 38 and HBV capsid protein (PDB:5T2P) |
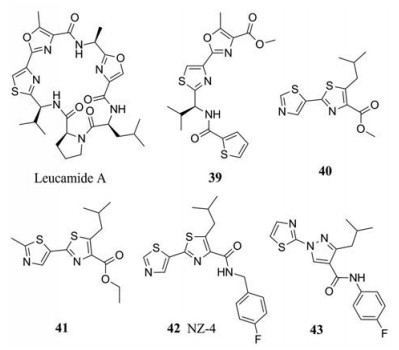
天然产物结构多样, 已成为发现新骨架药物的一条重要途径。其中, 杂环串联类的结构可以与多种靶标相互作用, 具有广泛的生物活性。Leucamide A是一种由环肽骨架构成的天然产物, 来源于澳大利亚海绵Leucetta microraphis, 可以抑制一些肿瘤细胞的生长, 但是抗病毒活性较弱[45]。
2008年, 左建平等[46]以leucamide A为先导化合物, 设计合成了一系列由甲基噁唑和噻唑组成的杂环串联类衍生物, 其中化合物39表现出中等的抗HBV活性(EC50 = 76.4 μmol·L-1), 是一类具有新骨架的HBV抑制剂。又继续以39为先导化合物, 设计合成了一系列4, 2-并杂环类衍生物, 其中串联双噻唑类抑制剂40活性较好, 细胞毒性较低(EC50 = 2.4 μmol·L-1, CC50 = 600 μmol·L-1); 在噻唑环上继续引入甲基(41)可以进一步提高活性, 但同时也增加了细胞毒性(EC50 = 0.14 μmol·L-1, CC50 = 118.2 μmol·L-1)。2014年, 该课题组报道了一个活性更好的分子42 (NZ-4), 其在HepG2.2.15细胞中抑制HBV DNA的EC50值达到了1.33 μmol·L-1, CC50为50.4 μmol·L-1; 机制研究表明NZ-4通过抑制pgRNA和HBcAg二聚体复合物的结合来干扰衣壳蛋白的组装, 但是不影响核衣壳的形成[47]。

|
2016年, 本课题组以NZ-4和AT-130为先导化合物, 采用骨架跃迁、分子杂合和生物电子等排的药物设计策略, 设计并合成了一批吡唑类衍生物。其中化合物43对HBsAg和HBeAg分泌的抑制活性分别为24.33和2.22 μmol·L-1, 有进一步研究的价值[48]。
作为一种从天然产物中演变而来抗HBV的新化学实体, 串联类抑制剂无疑具有较好的研发前景, 但是目前该类抑制剂与HBV衣壳蛋白的晶体复合物尚未被报道, 只能采用基于配体的药物设计策略。
5 二甲酰噻吩类抑制剂2019年, Huber等[49]通过高通量筛选发现了一个新型的二甲酰噻吩类HBV抑制剂44 (HepAD38细胞系, EC50 = 3.6 ± 0.2 μmol·L-1, CC50 > 300 μmol·L-1), 机制研究表明44使核心蛋白分布在细胞质中, 阻止其进入细胞核, 但不减少细胞内核心蛋白的总量。该课题组采取生物电子等排等策略对44进行结构优化, 合成了一批新型二甲酰噻吩类衍生物, 其中45 (EC50 = 0.11 ± 0.01 μmol·L-1, CC50 > 100 μmol·L-1)和46 (EC50 = 0.31 ± 0.07 μmol·L-1, CC50 > 100 μmol·L-1)的体外抗病毒活性大幅提升, 46在小鼠体内的药代动力学性质较好, 以10 mg·kg-1的剂量口服给药时, 生物利用度可达46%[50]。
|
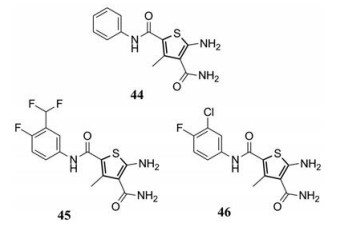
|
2015年, 左建平等[51]首次报道了一个以哒嗪酮为骨架的新型HBV衣壳蛋白抑制剂47, 其对细胞外和细胞内的HBV DNA的EC50值分别为1.5 ± 0.2和1.9 ± 0.1 μmol·L-1; 机制研究表明该化合物可以诱导不含病毒基因的核衣壳的组装, 并干扰核心蛋白的正常装配。2016年, Lu等[52]对47中的苯环进行了优化得到了活性更好的化合物48 (EC50 = 0.087 ± 0.002μmol·L-1, CC50 = 90.6 ± 2.06 μmol·L-1)。

|
近期, 两个HBV衣壳蛋白抑制剂ABI-H0731 (49)和AB-423 (50)进入了Ⅰ期临床试验, 但具体结构尚未公开。研究表明, ABI-H0731可以降低HBV核衣壳中pgRNA的含量; 其在HepAD38细胞和HBV感染的人原代肝细胞均表现了对HBV DNA复制的抑制活性, EC50值分别为0.37和0.15 μmol·L-1。AB-423可以抑制pgRNA的衣壳化, 还可以抑制核衣壳的脱壳来抑制cccDNA的合成, 其EC50值为260 nmol·L-1; 在临床前试验中, AB-423与RNAi药物ARB-1740联用在体外可以发挥对rcDNA的协同抑制作用, 在体内模型中也可协同减少HBV DNA和HBsAg的含量[53, 54]。
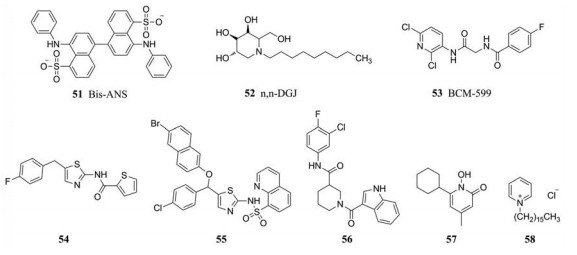
Zlotnick等[55]采用平衡透析法鉴定出新型的二磺酸联萘类HBV衣壳蛋白装配抑制剂Bis-ANS (51), 与核心蛋白二聚体结合后中断其继续组装, 产生一些非衣壳的聚合物。
Lu等[56]研究发现亚氨基糖类抑制剂n, n-DGJ (52)可以抑制HBV衣壳蛋白的成熟和破坏衣壳蛋白的稳定性, 是一种新型的HBV衣壳蛋白抑制剂。
为发现新结构的HBV衣壳蛋白抑制剂, Cho等[57]以Cp149模拟HBV衣壳蛋白, 通过计算机虚拟筛选得到一批苗头化合物; 进一步活性筛选发现化合物BCM-599 (53)具有较好的体外抗HBV活性, 其EC50为13 ± 1 μmol·L-1 (HepG2.2.15细胞系)。
2019年, Pan等[58]在体外模拟衣壳蛋白装配的环境, 通过虚拟筛选发现了一个具有抗HBV活性的单噻唑结构类似物54 (EC50 = 6.15 μmol·L-1, CC50 > 500 μmol·L-1), 并对其进行了构效关系研究得到了活性提高近35倍的化合物55 (EC50 = 0.18 μmol·L-1, CC50 > 50 μmol·L-1)。同年, Pei等[59]通过高通量筛选得到了一个骨架新颖的HBV抑制剂56 (HepAD38细胞系, EC50 = 0.02 μmol·L-1, CC50 ≈ 35 μmol·L-1; HepG2.2.15细胞系, EC50 = 0.05 μmol·L-1, CC50 ≈ 35 μmol·L-1), 在机制研究中发现56可以诱导不含病毒基因的“空壳”的形成, 与哒嗪酮类抑制剂作用机制相似。
8 通过药物再利用发现新型衣壳蛋白抑制剂除了经典的药物化学方法, 即“分子设计、合成、结构优化、活性测试”等步骤来发现药物, 对于已经上市的药物或者处于临床研究的候选药物进行新应用的二次开发, 可以发现其对于新靶标或者其他疾病的发展潜力。这种药物再利用的策略, 可以避开药物的安全性评价, 大大节省开发成本, 提高药物发现的效率。
在HBV衣壳蛋白小分子抑制剂探索过程中, 2019年Kang等[60]利用药物再利用策略对978种FDA已经批准上市的药物在HepG2.2.15细胞中对HBV DNA复制活性进行筛选, 发现化合物环吡酮57 (ciclopirox)有高效的HBV抗病毒活性。环吡酮是一种合成的抗真菌抑制剂, 研究发现, 它可以通过阻断HBV衣壳组装(IC50 = 445 ± 17 nmol·L-1), 强烈抑制细胞和小鼠中的HBV复制。
同年, Seo等[61]将含有抗菌活性的氯化十六烷基吡啶(cetylpyridinium chloride, CPC) 58对抗HBV活性进行测试, 并且确定CPC可以作为HBV的新型抑制剂, 抑制HBV核衣壳装配活性的IC50为2~3 μmol·L-1。利用CPC与核心蛋白的计算机模拟对接、CPC与病毒核衣壳结合的微型热泳分析、以及体外核衣壳形成测定, 他们发现CPC特异性地与二聚体病毒衣壳蛋白(核心蛋白或HBcAg)相互作用。与其他HBV抑制剂(如苯磺酰胺和磺胺)相比, CPC在HepG2.2.15细胞系中获得了明显更好的HBV颗粒数减少, CPC还可以抑制小鼠水动力模型系统中的HBV复制。总的来说, 该研究表明CPC可以抑制衣壳组装并导致HBV生物合成减少, 是一种可以减少HBV颗粒的有效分子。
9 总结与展望HBV感染宿主后难以被根除, 现有的核苷类药物无法满足患者的需求。为寻求能够治愈乙肝的手段, 科学家们将更多的目光投向作用于其他靶点的非核苷类HBV抑制剂。衣壳装配是HBV生命周期中的关键步骤, 随着结构生物学的发展, 对HBV核衣壳的形态构造和生物学功能有了全方位的认识。研究靶向于核心蛋白、干扰核衣壳的正常组装的小分子药物成为了一大热点。
|
在HBV衣壳蛋白抑制剂的发展初始阶段, 根据已报道的衣壳蛋白抑制剂的化学结构, 采用基于配体的药物设计策略, 如生物电子等排、骨架跃迁、优势结构片段、分子杂合、环化-构象保持、并环等, 这些策略的灵活和综合的应用完善了同一类型骨架的构效关系, 有助于进一步提高抗病毒活性、改善药物成药性。随着结构生物学的发展, 衣壳蛋白与配体的晶体复合物被解析, 采用基于结构的合理药物设计, 靶向于关键氨基酸形成额外相互作用力, 如氢键、疏水作用力等, 则可以更高效地发现高活性、高亲和力、抗耐药性的先导化合物; 除了传统的优化先导化合物的方法, 将药物化学与生物信息学和人工智能技术结合, 对已上市或者未批准的药物进行高通量表型筛选、反向药效团匹配虚拟筛选等方法探索药物新适应症, 这种药物再利用策略用以发现药物的新应用也是一种高效率的药物发现方法; 不容忽视的是, 来自于动物、植物、昆虫、微生物、海洋生物等的结构多样的天然产物也是HBV衣壳蛋白新骨架抑制剂的一个重要来源。
除衣壳蛋白抑制剂外, 靶向于其他HBV生命周期重要靶点的抑制剂也是近年来乙肝领域的热点之一。随着新靶标的发现及筛选方法的建立, 将会有更多的高活性及成药性小分子被报道, 为新一代非核苷类HBV药物的上市奠定基础。在科技发展如此迅速、学科交叉融合的新时代背景下, 将有望根治HBV感染。
| [1] |
WHO. Fact sheets of HBV [EB/OL]. WHO, 2019, [2020-03-02].https://www.who.int/zh/news-room/fact-sheets/detail/hepatitis-b.
|
| [2] |
Böttcher B, Wynne SA, Crowther RA. Determination of the fold of the core protein of hepatitis B virus by electron cryomicroscopy[J]. Nature, 1997, 386: 88-91. DOI:10.1038/386088a0 |
| [3] |
Ceres P, Zlotnick A. Weak protein-protein interactions are sufficient to drive assembly of hepatitis B virus capsids[J]. Biochemistry, 2002, 41: 11525-11531. DOI:10.1021/bi0261645 |
| [4] |
Katen SP, Chirapu SR, Finn MG, et al. Trapping of hepatitis B virus capsid assembly intermediates by phenylpropenamide assembly accelerators[J]. ACS Chem Biol, 2010, 5: 1125-1136. DOI:10.1021/cb100275b |
| [5] |
Zhou Z, Hu T, Zhou X, et al. Heteroaryldihydropyrimidine (HAP) and sulfamoylbenzamide (SBA) inhibit hepatitis B virus replication by different molecular mechanisms[J]. Sci Rep, 2017, 7: 42374. DOI:10.1038/srep42374 |
| [6] |
Nair S, Li L, Francis S, et al. Use of a fluorescent analogue of a HBV core protein-directed drug to interrogate an antiviral mechanism[J]. J Am Chem Soc, 2018, 140: 15261-15269. DOI:10.1021/jacs.8b07988 |
| [7] |
Schlicksup CJ, Wang JC, Francis S, et al. Hepatitis B virus core protein allosteric modulators can distort and disrupt intact capsids[J]. ELife, 2018, 7: e31473. DOI:10.7554/eLife.31473 |
| [8] |
King RW, Ladner SK, Miller TJ, et al. Inhibition of human hepatitis B virus replication by AT-61, a phenylpropenamide derivative, alone and in combination with (-)beta-L-2', 3'-dideoxy-3'-thiacytidine[J]. Antimicrob Agents Chemother, 1998, 42: 3179-3186. DOI:10.1128/AAC.42.12.3179 |
| [9] |
Perni RB, Conway SC, Ladner SK, et al. Phenylpropenamide derivatives as inhibitors of hepatitis B virus replication[J]. Bioorg Med Chem Lett, 2000, 10: 2687-2690. DOI:10.1016/S0960-894X(00)00544-8 |
| [10] |
Dong WL, Liu ZX, Liu XH, et al. Synthesis and antiviral activity of new acrylamide derivatives containing 1, 2, 3-thiadiazole as inhibitors of hepatitis B virus replication[J]. Eur J Med Chem, 2010, 45: 1919-1926. |
| [11] |
Wang P, Naduthambi D, Mosley RT, et al. Phenylpropenamide derivatives: anti-hepatitis B virus activity of the isomer, SAR and the search for novel analogs[J]. Bioorg Med Chem Lett, 2011, 21: 4642-4647. DOI:10.1016/j.bmcl.2011.05.077 |
| [12] |
Katen SP, Tan Z, Chirapu SR, et al. Assembly-directed antivirals differentially bind quasiequivalent pockets to modify hepatitis B virus capsid tertiary and quaternary structure[J]. Structure, 2013, 21: 1406-1416. DOI:10.1016/j.str.2013.06.013 |
| [13] |
Zlotnick A, Venkatakrishnan B, Tan ZN, et al. Core protein: a pleiotropic keystone in the HBV lifecycle[J]. Antiviral Res, 2015, 121: 82-93. |
| [14] |
Liu N, Zhao FB, Jia HY, et al. Non-nucleoside anti-HBV agents: advances in structural optimization and mechanism of action investigations[J]. Med Chem Commun, 2015, 6: 521-535. DOI:10.1039/C4MD00521J |
| [15] |
Weber O, Schlemmer KH, Hartmann E, et al. Inhibition of human hepatitis B virus (HBV) by a novel non-nucleosidic compound in a transgenic mouse model[J]. Antiviral Res, 2002, 54: 69-78. DOI:10.1016/S0166-3542(01)00216-9 |
| [16] |
Deres K, Schröder CH, Paessens A, et al. Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids[J]. Science, 2003, 299: 893-896. DOI:10.1126/science.1077215 |
| [17] |
Stray SJ, Bourne CR, Punna S, et al. A heteroaryldihydropyrimidine activates and can misdirect hepatitis B virus capsid assembly[J]. Proc Natl Acad Sci U S A, 2005, 102: 8138-8143. |
| [18] |
Bourne CR, Finn MG, Zlotnick A. Global structural changes in hepatitis B virus capsids induced by the assembly effector HAP1[J]. J Virol, 2006, 80: 11055-11061. DOI:10.1128/JVI.00933-06 |
| [19] |
Bourne C, Lee S, Venkataiah B, et al. Small-molecule effectors of hepatitis B virus capsid assembly give insight into virus life cycle[J]. J Virol, 2008, 82: 10262-10270. DOI:10.1128/JVI.01360-08 |
| [20] |
Zhu X, Zhao G, Zhou X, et al. 2, 4-Diaryl-4, 6, 7, 8-tetrahydroquinazolin-5(1H)-one derivatives as anti-HBV agents targeting at capsid assembly[J]. Bioorg Med Chem Lett, 2010, 20: 299-301. DOI:10.1016/j.bmcl.2009.10.119 |
| [21] |
Yang XY, Xu XQ, Guan H, et al. A new series of HAPs as anti-HBV agents targeting at capsid assembly[J]. Bioorg Med Chem Lett, 2014, 24: 4247-4249. DOI:10.1016/j.bmcl.2014.07.032 |
| [22] |
Boucle S, Lu X, Bassit L, et al. Synthesis and antiviral evaluation of novel heteroarylpyrimidines analogs as HBV capsid effectors[J]. Bioorg Med Chem Lett, 2017, 27: 904-910. DOI:10.1016/j.bmcl.2017.01.010 |
| [23] |
Tu J, Li JJ, Shan ZJ, et al. Exploring the binding mechanism of heteroaryldihydropyrimidines and hepatitis B virus capsid combined 3D-QSAR and molecular dynamics[J]. Antiviral Res, 2017, 137: 151-164. DOI:10.1016/j.antiviral.2016.11.026 |
| [24] |
Grimm D, Thimme R, Blum HE. HBV life cycle and novel drug targets[J]. Hepatol Int, 2011, 5: 644-653. DOI:10.1007/s12072-011-9261-3 |
| [25] |
Stray SJ, Zlotnick A. BAY 41-4109 has multiple effects on hepatitis B virus capsid assembly[J]. J Mol Recognit, 2006, 19: 542-548. DOI:10.1002/jmr.801 |
| [26] |
Wu G, Liu B, Zhang Y, et al. Preclinical characterization of GLS4, an inhibitor of hepatitis B virus core particle assembly[J]. Antimicrob Agents Chemother, 2013, 57: 5344-5354. DOI:10.1128/AAC.01091-13 |
| [27] |
Klumpp K, Lam AM, Lukacs C, et al. High-resolution crystal structure of a hepatitis B virus replication inhibitor bound to the viral core protein[J]. Proc Natl Acad Sci U S A, 2015, 112: 15196-15201. DOI:10.1073/pnas.1513803112 |
| [28] |
Zhou X, Li L, Deng P, et al. Characterization of metabolites of GLS4 in humans using ultrahigh-performance liquid chromatography/quadrupole time-of-flight mass spectrometry[J]. Rapid Commun Mass Spectrom, 2013, 27: 2483-2492. DOI:10.1002/rcm.6710 |
| [29] |
Qiu Z, Lin X, Zhou M, et al. Design and synthesis of orally bioavailable 4-methyl heteroaryldihydropyrimidine based hepatitis B virus (HBV) capsid inhibitors[J]. J Med Chem, 2016, 59: 7651-7666. DOI:10.1021/acs.jmedchem.6b00879 |
| [30] |
Qiu Z, Lin X, Zhang W, et al. Discovery and pre-clinical characterization of third-generation 4-H heteroaryldihydropyrimidine (HAP) analogues as hepatitis B virus (HBV) capsid inhibitors[J]. J Med Chem, 2017, 60: 3352-3371. DOI:10.1021/acs.jmedchem.7b00083 |
| [31] |
Li X, Zhou K, He H, et al. Design, synthesis, and evaluation of tetrahydropyrrolo [1, 2-c] pyrimidines as capsid assembly inhibitors for HBV treatment[J]. ACS Med Chem Lett, 2017, 8: 969-974. DOI:10.1021/acsmedchemlett.7b00288 |
| [32] |
Liu X, Jia H, Zhan P, et al. Preparation of dihydropyrimidine-triazole derivatives useful as anti-HBV agents: CN, 107501257 [P]. 2017-12-22.
|
| [33] |
Ren Q, Liu X, Yan G, et al. 3-((R)-4-(((R)-6-(2-bromo-4-fluorophenyl)-5-(ethoxycarbonyl)-2-(thiazol-2-yl)-3, 6-dihydropyrimidin-4-yl)methyl)morpholin-2-yl)propanoic acid (HEC72702), a novel hepatitis B virus capsid inhibitor based on clinical candidate GLS4[J]. J Med Chem, 2018, 61: 1355-1374. |
| [34] |
Guo L, Hu T, Hu Y, et al. Preparation of heteroaryldihydropyrimidine derivatives for use in the treatment of hepatitis B virus infection: WO, 2014184328 [P]. 2014-11-20.
|
| [35] |
Guo L, Hu T, Kou B, et al. Preparation of novel 6-fused heteroaryldihydropyrimidines for the treatment and prophylaxis of hepatitis B virus (HBV) infection: CN, 107513073 [P]. 2017-12-26.
|
| [36] |
Ren QY, Liu XC, Huang JZ, et at. 1. Dihydropyrimidine compound and uses of dihydropyrimidine compound in drugs: CN, 109111451 [P]. 2019-01-01.
|
| [37] |
Campagna MR, Liu F, Mao R, et al. Sulfamoylbenzamide derivatives inhibit the assembly of hepatitis B virus nucleocapsids[J]. J Virol, 2013, 87: 6931-6942. DOI:10.1128/JVI.00582-13 |
| [38] |
Sari O, Boucle S, Cox BD, et al. Synthesis of sulfamoylbenzamide derivatives as HBV capsid assembly effector[J]. Eur J Med Chem, 2017, 138: 407-421. DOI:10.1016/j.ejmech.2017.06.062 |
| [39] |
Wu S, Zhao Q, Zhang P, et al. Discovery and mechanistic study of benzamide derivatives that modulate hepatitis B virus capsid assembly[J]. J Virol, 2017, 91: e00519-17. |
| [40] |
Vandyck K, Rombouts G, Stoops B, et al. Synthesis and evaluation of N-phenyl-3-sulfamoyl-benzamide derivatives as capsid assembly modulators inhibiting hepatitis B virus (HBV)[J]. J Med Chem, 2018, 61: 6247-6260. DOI:10.1021/acs.jmedchem.8b00654 |
| [41] |
Wang Z, Fan G, Lu C, et al. Bicyclic nucleocapsid inhibitors and use of same as drugs in treatment of hepatitis B: WO, 2018202155 [P]. 2018-11-08.
|
| [42] |
Lam AM, Espiritu C, Vogel R, et al. Preclinical characterization of NVR 3-778, a first-in-class capsid assembly modulator against hepatitis B virus[J]. Antimicrob Agents Chemother, 2019, 63: e01734-18. |
| [43] |
An efficacy, safety, and pharmacokinetics study of JNJ-56136379 in participants with chronic hepatitis B virus infection [EB/OL]. 2019 [2019-10-2].https://ClinicalTrials.gov/show/NCT03361956.
|
| [44] |
In capsid assembly modulator JNJ-56136379 prevents de novo infection of primary human hepatocytes with hepatitis B virus [C] // American Association for the Study of Liver Disease. Boston: The Liver Meeting, 2016.
|
| [45] |
Kehraus S, König GM, Wright AD, et al. Leucamide A: a new cytotoxic heptapeptide from the Australian sponge Leucettamicroraphis[J]. J Org Chem, 2002, 67: 4989-4992. DOI:10.1021/jo020058r |
| [46] |
Chen HJ, Wang WL, Wang GF, et al. Rational design and synthesis of 2, 2-bisheterocycle tandem derivatives as non-nucleoside hepatitis B virus inhibitors[J]. ChemMedChem, 2008, 3: 1316-1321. |
| [47] |
Yang L, Shi LP, Chen HJ, et al. Isothiafludine, a novel non-nucleoside compound, inhibits hepatitis B virus replication through blocking pregenomic RNA encapsidation[J]. Acta Pharmacol Sin, 2014, 35: 410-418. DOI:10.1038/aps.2013.175 |
| [48] |
Jia H, Bai F, Liu N, et al. Design, synthesis and evaluation of pyrazole derivatives as non-nucleoside hepatitis B virus inhibitors[J]. Eur J Med Chem, 2016, 123: 202-210. DOI:10.1016/j.ejmech.2016.07.048 |
| [49] |
Huber AD, Pineda DL, Liu D, et al. Novel hepatitis B virus capsid-targeting antiviral that aggregates core particles and inhibits nuclear entry of viral cores[J]. ACS Infect Dis, 2019, 5: 750-758. DOI:10.1021/acsinfecdis.8b00235 |
| [50] |
Tang J, Huber AD, Pineda DL, et al. 5-Aminothiophene-2, 4-dicarboxamide analogues as hepatitis B virus capsid assembly effectors[J]. Eur J Med Chem, 2019, 164: 179-192. DOI:10.1016/j.ejmech.2018.12.047 |
| [51] |
Wang YJ, Lu D, Xu YB, et al. A novel pyridazinone derivative inhibits hepatitis B virus replication by inducing genome-free capsid formation[J]. Antimicrob Agents Chemother, 2015, 59: 7061-7072. DOI:10.1128/AAC.01558-15 |
| [52] |
Lu D, Liu F, Xing W, et al. Optimization and synthesis of pyridazinone derivatives as novel inhibitors of hepatitis B virus by inducing genome-free capsid formation[J]. ACS Infect Dis, 2017, 3: 199-205. DOI:10.1021/acsinfecdis.6b00159 |
| [53] |
Huang Q, Zong YH, Mercier A, et al. Blockage of HBV virus replication and inhibition of cccDNA establishment by core protein allosteric modifiers (CpAMs)[J]. Hepatology, 2016, 64: 937A-938A. |
| [54] |
Mani N, Cole AG, Ardzinski A, et al. The HBV capsid inhibitor AB-423 exhibits a dual mode of action and displays additive/synergistic effects in in vitro combination studies[J]. Hepatology, 2016, 64: 123A-124A. |
| [55] |
Zlotnick A, Ceres P, Singh S, et al. A small molecule inhibits and misdirects assembly of hepatitis B virus capsids[J]. J Virol, 2002, 76: 4848-4854. DOI:10.1128/JVI.76.10.4848-4854.2002 |
| [56] |
Lu X, Tran T, Simsek E, et al. The alkylated imino sugar, n-(n-nonyl)-deoxygalactonojirimycin, reduces the amount of hepatitis B virus nucleocapsid in tissue culture[J]. J Virol, 2003, 77: 11933-11940. DOI:10.1128/JVI.77.22.11933-11940.2003 |
| [57] |
Cho MH, Jeong H, Kim YS, et al. 2-Amino-N-(2, 6-dichloropyridin-3-yl)acetamide derivatives as a novel class of HBV capsid assembly inhibitor[J]. J Viral Hepat, 2014, 21: 843-852. DOI:10.1111/jvh.12214 |
| [58] |
Pan T, Ding Y, Wu L, et al. Design and synthesis of aminothiazole based hepatitis B virus (HBV) capsid inhibitors[J]. Eur J Med Chem, 2019, 166: 480-501. |
| [59] |
Pei Y, Wang C, Ben H, et al. Discovery of new hepatitis B virus capsid assembly modulators by an optimal high-throughput cell-based assay[J]. ACS Infect Dis, 2019, 5: 778-787. DOI:10.1021/acsinfecdis.9b00030 |
| [60] |
Kang JA, Kim S, Park M, et al. Ciclopirox inhibits hepatitis B virus secretion by blocking capsid assembly[J]. Nat Commun, 2019, 10: 2184. DOI:10.1038/s41467-019-10200-5 |
| [61] |
Seo HW, Seo JP, Cho Y, et al. Cetylpyridinium chloride interaction with the hepatitis B virus core protein inhibits capsid assembly[J]. Virus Res, 2019, 263: 102-111. DOI:10.1016/j.virusres.2019.01.004 |