 2020, Vol. 55
2020, Vol. 55

2. 山东省中比抗病毒创新药物合作研究中心, 山东 济南 250012;
3. 山东中医药大学药学院, 山东 济南 250355;
4. 美国伊利诺伊大学芝加哥校区医学院微生物学与免疫学系, 伊利诺伊州 芝加哥 60612

 , RONG Li-jun4, LIU Xin-yong1,2
, RONG Li-jun4, LIU Xin-yong1,2
2. China-Belgium Collaborative Research Center for Innovative Antiviral Drugs of Shandong Province, Jinan 250012, China;
3. College of Pharmacy, Shandong University of Traditional Chinese Medicine, Jinan 250355, China;
4. Department of Microbiology and Immunology, College of Medicine, University of Illinois at Chicago, Chicago, IL 60612, USA
冠状病毒(coronavirus, CoVs)是一类常见的病毒大家族, 在自然界中广泛存在, 可感染人类并容易引起急慢性呼吸道系统疾病[1]。该属病毒的广泛宿主特点以及自身基因组的结构特征使其在进化过程中极易发生基因重组和变异, 呈现遗传多样性。冠状病毒科分为α、β、γ、δ 4个属, 已知感染人的冠状病毒共有7种(表 1), 其中前4种在人群中较为常见(HCoV-229E、HCoV-OC43、HCoV-NL63、HCoV-HKU1), 致病性较低, 一般仅引起类似普通感冒的轻微呼吸道症状。另外3种对人类健康具有严重的威胁, 包括2003年暴发的严重急性呼吸综合征冠状病毒(severe acute respiratory syndrome coronavirus, SARS-CoV)、2012年暴发的中东呼吸综合征病毒(Middle East respiratory syndrome coronavirus, MERS-CoV)和最近暴发的新型冠状病毒(SARS-CoV-2)[2-5], 由SARS-CoV-2导致的疾病被命名为COVID-19 (coronavirus disease 2019)[6]。
| Table 1 Seven members of human coronavirus |
目前COVID-19已在世界大部分国家和地区扩展和蔓延, 确诊与死亡人数在不断增加。由于SARS-CoV-2具有高传染性及高致死率, 对于人类健康和社会公共卫生安全产生了极大的威胁。新型冠状病毒感染的肺炎诊疗方案(试行第五版)推荐抗病毒治疗可试用α型干扰素(广谱抗病毒药物)雾化吸入、洛匹那韦(lopinavir, LPV)/利托那韦(ritonavir, RTV) (HIV蛋白酶抑制剂)或可加用利巴韦林(广谱抗病毒药物)。可以看出, 对包括SARS-CoV-2在内的所有人类冠状病毒感染, 除了对症治疗外, 目前尚无确切有效的特异性治疗药物或疫苗[7, 8], 因此研发安全有效、具有临床应用价值的抗冠状病毒药物刻不容缓。
药物化学是基于靶标的药物发现的重要手段, 采用先进合理的药物化学策略可提高药物发现的成功率。因此, 笔者结合专业特长, 从药物化学的视角, 精选抗SARS-CoV、MERS-CoV等冠状病毒药物的研究案例, 总结了药物发现的药物化学策略。并针对新近暴发的SARS-CoV-2感染疫情, 及时分析了药物研究新动向, 提出了未来研究的新思路。
1 冠状病毒简介冠状病毒是一类表面具有包膜的球状病毒, 直径为120~160 nm[9]。病毒粒子的外周是整齐排列的纤突, 被脂质膜所包裹, 在脂质膜上含有3种糖蛋白。分别是刺突糖蛋白(spike glycoprotein, S蛋白), 负责与宿主受体的粘附; 不同冠状病毒的刺突蛋白不一样, 相应的宿主细胞上的受体也就不同, 这种特异的结合决定了病毒可以感染的细胞类型以及能够入侵的物种范围。膜蛋白(membrane protein, M蛋白), 塑造了病毒粒子的形状; 以及少量的包膜蛋白(envelope protein, E蛋白), 促进病毒的组装和释放。病毒的基因组RNA缠绕在核衣壳蛋白(nucleocapsid, N蛋白)上形成螺旋形核衣壳(图 1A)。

|
Figure 1 Structural coronavirus (A) and genetic structure of SARS and MERS virus (B) |
冠状病毒RNA基因组是不分段的单股正链RNA, 大小约为26~32 kb, 是RNA病毒中基因组最大的病毒[10]。冠状病毒的5'端存在甲基化的帽子、3'端存在poly A尾巴, 与真核细胞的mRNA相似。病毒基因组的三分之二为开放阅读框1a/b (open reading frame, ORF1a/b), 编码两个复制酶多聚蛋白1a(pp1a)和pp1b。这些多聚蛋白被类木瓜蛋白酶(papain-like protease, PLpro)和类3C蛋白酶(3CLpro, 也称主蛋白酶, Mpro)切割, 产生非结构蛋白(功能性多肽), 如RNA依赖的RNA聚合酶(RNA-dependent RNA polymerase, RdRp)和解旋酶(helcase, Hel)参与病毒RNA的转录与复制。其后的三分之一基因分别编码S、E、M和N结构蛋白(图 1B)。由于细胞中修复DNA复制错误的机制并不会修复病毒RNA, 因此RNA病毒基因的复制过程经常出错。再加上冠状病毒的基因组比较长(由3万个碱基组成), 病毒在复制时需要的碱基也就越多, 出错的几率就会更大。因此, 这些冠状病毒极易出现变异性[11]。
冠状病毒的复制周期包括侵入、复制、组装与分泌等阶段(图 2)[3]。首先病毒S刺突糖蛋白的S1亚基上的受体结合域(receptor binding domain, RBD)与宿主细胞表面的受体结合, 触发S2亚基的构象变化, 从而诱导病毒和细胞膜的融合[12, 13]。冠状病毒通过内体途径或细胞表面非内体途径进入宿主细胞, 低pH和pH依赖性的半胱氨酸蛋白酶、组织蛋白酶可以促进冠状病毒通过内体途径进入宿主细胞[7]。然后, 病毒将遗传物质以内吞的方式释放到宿主细胞质中, RNA作为mRNA翻译生成RdRp。进而以RNA基因组为模板在RdRp的作用下生成负链RNA, 生成的负链RNA进一步复制形成新的正链RNA。此外, 亚基因组mRNA在这一过程中被合成, 再被翻译形成其他结构蛋白。最后, 核衣壳蛋白(N)和基因组RNA组装形成螺旋核衣壳, 并与其他结构蛋白装配形成病毒颗粒, 经胞吐释放到细胞外。理论上, 阻断病毒复制周期的任何一个环节, 都可以抑制病毒复制。

|
Figure 2 Life cycle diagram of coronavirus |
目前, 文献所报道的抗冠状病毒药物靶点主要包括:病毒配体S蛋白、与病毒转录复制相关的RdRp、PLpro和3CLpro, 以及宿主因子血管紧张素转化酶2 (angiotensin-converting enzyme 2, ACE2)等[14, 15]。
2 冠状病毒抑制剂先导化合物发现的药物化学策略先导化合物的发现是药物研发的关键环节, 也是药物化学研究的核心任务之一。当前, SARS-CoV、MERS-CoV等冠状病毒抑制剂的先导化合物的发现途径主要包括:老药新用、基于靶标结构的虚拟筛选、基于病毒蛋白/酶的高通量筛选、基于细胞水平的表型筛选、基于靶标相似性的精准药物设计、基于靶标的全新药物设计等。先导化合物发现以后, 其结构优化主要包括一些常规药物化学策略[16, 17], 本文仅简明介绍, 着重论述抗冠状病毒先导化合物发现的途径(图 3), 为预防新疫情的药物研发提供参考。

|
Figure 3 Schematic diagram of medicinal chemistry strategies used in previous anti-coronavirus drug research |
老药新用又称药物再利用(drug repurposing), 是一种对已知药物(包括已经上市和未获批准的药物)的应用进行重新探索和开发, 并将其用于治疗其他疾病的新药研发策略。“老药”相比于全新结构药物, 其合成工艺成熟、安全性和药代动力学数据已知, 可以避免由ADMET引起的临床高失败率, 大大降低研发成本, 缩短上市时间。当然, “老药”其实不仅限于现有已上市药物, 还应该包含曾/现处于临床前或临床研究中的潜在候选物。近年来, 老药新用作为药物开发策略越来越受到重视[18], 特别是在新疫情暴发时, 成为最高效快捷的途径。广谱抗病毒药物是老药新用的首选, 此外还包括老药新用理念指导下的优势结构再定位。
2.1.1 广谱抗病毒药物的老药新用在长期治疗过程中, 病毒会通过自身突变产生耐药性。同时, 由于不同病毒编码的蛋白不同, 传统抗病毒药物对新发病毒不能起到及时、有效的治疗效果。广谱抗病毒药物可作用于多种病毒或同一种病毒的多种基因型, 具有“一石多鸟”之效。因此, 寻找安全有效的广谱抗病毒药物逐渐成为抗病毒领域的研究热点。
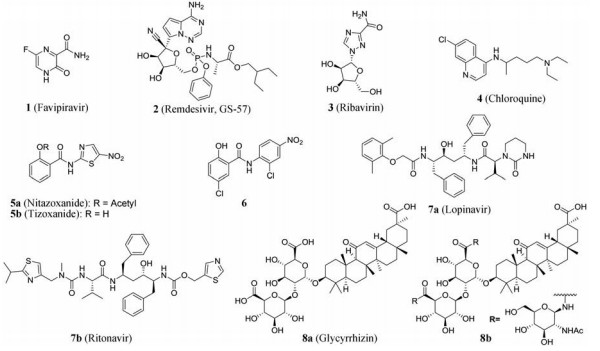
法匹拉韦(1, favipiravir)是由日本富山化工制药公司对一种吡嗪类似物进行化学修饰而发现的, 属于RNA聚合酶抑制剂, 具有广谱抗病毒活性[19]。法匹拉韦于2014年3月在日本获批上市, 最初用于治疗新型和复发型流感。随后研究表明法匹拉韦对多种RNA病毒均具有良好的抑制作用(如埃博拉病毒、诺罗病毒、狂犬病毒等)。法匹拉韦作为前药, 在真核生物磷酸核糖转移酶(hypoxanthine guanine phosphoribosyl transferase, HGPRT)的作用下转化为呋喃糖基5'-三磷酸代谢产物(favipiravir-RTP)。Favipiravir-RTP可掺入到病毒新合成的RNA链中, 并可抑制甲型流感病毒聚合酶的生理功能[20]。但其广谱抗RNA病毒活性的确切作用机制尚未完全阐明, 目前推测favipiravir-RTP可能会错误地掺入正在增长的病毒RNA链中, 或者通过与保守的聚合酶结构域结合而阻止病毒RNA复制[19]。2020年2月15日, 法匹拉韦获得国家药监局批准上市, 用于治疗流感[21]。国家卫健委曾在新闻发布会上指出法匹拉韦对SRAS-CoV-2具有抑制作用[22]。据中国科学院武汉病毒所和军事医学科学院毒物药物研究所的体外细胞(Vero E6)活性测定结果可知, 法匹拉韦对SRAS-CoV-2的抑制活性较低(EC50 = 61.88 μmol·L-1, CC50 > 400 μmol·L-1, SI > 6.46), 但由于该药之前对埃博拉病毒的抑制效果较好, 仍值得进一步研究[23]。目前正在进行治疗SARS-CoV-2的临床试验[22]。瑞德西韦(2, remdesivir, GS-5734)是吉利德科学公司开发的核苷类抗病毒药物, 是一种新型的腺苷类似物单磷酸酰胺前药, 在细胞内可转变成药理学活性形式三磷酸核苷(NTP), 通过抑制RdRp, 最终抑制病毒核酸的合成。瑞德西韦最初用于埃博拉(Ebola)病毒感染, 目前在刚果(金)开展治疗埃博拉出血热的Ⅱ和Ⅲ期临床研究。后续研究发现其具有广谱抗病毒活性, 在体外对呼吸道合胞病毒(respiratory syncytial virus, RSV)、鸠宁病毒(Junin virus)、拉沙热病毒(Lassa fever virus)、MERS-CoV、SARS-CoV以及尼帕病毒(Nipah virus)等多种RNA病毒均具有抑制活性, 这对瑞德西韦临床治疗SARS-CoV-2感染具有重要启示[24]。中国科学院武汉病毒所和军事医学科学院毒物药物研究所的体外细胞(Vero E6)活性测定结果表明瑞德西韦对SARS-CoV-2具有较优的抑制活性(EC50 = 0.77 μmol·L-1, CC50 > 100 μmol·L-1, SI > 129.87), 为新型冠状病毒肺炎的治疗带来了新的希望[23]。2020年1月31日《新英格兰医学杂志》报道了美国一例新型冠状病毒感染肺炎患者静脉注射瑞德西韦后症状迅速改善的病例报告, 虽然只是个例, 但能初步证明瑞德西韦在人体中不会引起较大的不良反应[25, 26]。目前, 瑞德西韦治疗SARS-CoV-2的Ⅲ期临床试验正在进行, 总样本量270例, 入组轻、中度新冠肺炎患者, 预期在4月27日结束[27]。利巴韦林(3, ribavirin)是一种具有广谱抗病毒活性的鸟苷类似物, 临床上用于治疗严重呼吸道合胞病毒感染、HCV感染和病毒性出血热[28]。自2003年SARS事件暴发以来, 对利巴韦林的研究不断, 但研究者发现利巴韦林在不同的细胞系中抑制SARS病毒的活性差别较大[29, 30]。2005, Morgenstern等[31]系统测定了利巴韦林在5种不同细胞系中的抗SARS-CoV的活性, 发现利巴韦林在猴Vero细胞中EC50 > 1 000 μg·mL-1, 在猪肾脏细胞系PK-15中的EC50值为2.2 μg·mL-1, 这种差距可能与利巴韦林抑制病毒复制的作用机制有关。2020年1月27日, 沈阳药科大学、华中科技大学等[32]根据SARS-CoV PLpro的结构, 采用同源建模方法模拟了SARS-CoV-2 PLpro的蛋白质结构, 运用基于蛋白结构的虚拟筛选, 从药物库(2 924个小分子化合物)、实验室自建的中药和天然产物数据库(约1 000个分子)中得到33个潜在的SARS-CoV-2 PLpro抑制剂, 其中包括利巴韦林, 预计对SARS-CoV-2有抑制作用。应注意的是, 利巴韦林属于鸟苷类似物, 可竞争性地抑制病毒的三磷酸鸟苷合成, 而此次筛选是以SARS-CoV-2 PLpro的蛋白质结构为靶点来进行的, 这种差异性值得进一步深入研究。
氯喹(4, chloroquine)是靶向宿主(溶酶体)的广谱抗病毒药物对免疫缺陷病毒、寨卡病毒、登革热病毒和冠状病毒均具有抑制活性[33, 34]。氯喹最初由德国科学家根据天然奎宁的化学结构设计而成, 是治疗疟疾的首选药物[35]。作为弱碱性的物质, 氯喹可以诱导宿主细胞内酸性细胞器(包括内吞体、溶酶体和高尔基体小泡)的pH升高。这种pH升高主要通过两种机制产生抗病毒作用: ①对在低pH下通过内吞体途径侵入细胞的病毒感染具有显著的抑制作用; ②可抑制病毒包膜糖蛋白在高尔基体和内质网内的蛋白酶和糖基转移酶的修饰, 破坏病毒包膜成熟而降低病毒的感染性。此外, 氯喹还具有免疫调节和抗炎等多种作用[36, 37]。中国科学院的细胞活性(Vero E6)测定结果表明, 氯喹对SARS-CoV-2具有较优的抑制活性(EC50 = 1.13 μmol·L-1, CC50 > 100 μmol·L-1, SI > 88.50), 目前氯喹治疗SARS-CoV-2的临床试验正在进行[23, 38]。
硝唑尼特(5a, nitazoxanide, NTZ)是一种噻唑类化合物, 2002年在美国上市, 用于治疗贾第鞭毛虫或隐孢子虫引起的腹泻[39, 40]。最初, NTZ是在氯硝柳胺(6, niclosamide)的结构上改造而来(以硝基噻唑取代间硝基氯苯), 被开发为一种口服抗寄生虫剂[41]。NTZ口服吸收之后在血液中迅速脱乙酰化, 形成活性代谢物替唑尼特(5b, tizoxanide, TIZ)。实验室以及动物模型实验的研究发现, NTZ和TIZ对多种流感病毒、A72细胞内的犬冠状病毒复制(EC50 = 1.0 ng·mL-1)和小鼠冠状病毒、小鼠肝炎病毒A59 (MHV-A59)、牛冠状病毒L9株(BCoV-L9)和人肠道冠状病毒4408株(HECoV-4408)的N蛋白的表达均有抑制作用(IC50约为0.3 μg·mL-1)[41-44]。此外, NTZ还可抑制促炎性细胞因子的过量释放, 促进成纤维细胞产生干扰素α和干扰素β并激活PKR诱导宿主先天性免疫应答[44, 45]。据中国科学院武汉病毒所和军事医学科学院毒物药物研究所的体外细胞(Vero E6)活性测定结果可知NTZ可在低微摩尔浓度下(EC50 = 2.12 μmol·L-1, CC50 > 35.53 μmol·L-1, SI > 16.76)抑制SARS-CoV-2, 是一种具有抗病毒潜力的商用抗冠状病毒制剂[23]。此外, 研究表明氯硝柳胺也能抑制SARS-CoV的复制。免疫印迹分析显示, 在1.56 μmol·L-1的氯硝柳胺浓度下, 病毒抗原合成可被完全抑制, 可见这类结构的化合物值得进一步研究[46]。洛匹那韦/利托那韦(7, lopinavir/ritonavir, LPV/r, 商品名:克力芝)于2000年被美国FDA批准上市用于艾滋病的抗病毒治疗。克力芝的主要成分是洛匹那韦(lopinavir), 通过和病毒的蛋白酶结合来抑制其活性, 进而使得病毒复制后期的Gag-Pol多聚蛋白不能顺利裂解, 产生未成熟且感染能力减弱的病毒[47]。冠状病毒的复制也需要病毒蛋白酶的作用, 因此, 洛匹那韦/利托那韦可能会与冠状病毒蛋白酶结合从而抑制其正常功能。虽然前期体外研究表明, 洛匹那韦/利托那韦和干扰素β对MERS-CoV的抑制活性不佳, 但在SARS-CoV实验中具有治疗作用[48-51]。国家卫生健康委颁布的《新型冠状病毒感染的肺炎诊疗方案》试行版本中, 已指出克力芝可试用于新型冠状病毒的抗病毒治疗[47]。目前克力芝治疗SARS-CoV-2的临床试验正在进行[52]。
干扰素可以促进人体抗病毒蛋白的生成, 具有抗病毒、抗肿瘤和免疫调节的作用。体外实验表明, 干扰素对冠状病毒的复制具有抑制作用, 其中干扰素β的抑制能力最强。临床上, 将干扰素与利巴韦林的联合使用可显著抑制冠状病毒的复制, 但由于较严重的不良反应以及患者的依从性差等缺点, 其疗效并不稳定[53-55]。
甘草酸(8a, glycyrrhizin)是一种从甘草的根和根茎中分离出来的三萜, 对多种病毒(如疱疹病毒、人类免疫缺陷病毒等)都具有活性。临床上用于治疗肝炎、支气管炎、胃溃疡、艾滋病、某些癌症和皮肤病[56]。2003年, SARS病毒暴发。Cinatl等[57]通过细胞水平的活性筛选发现甘草酸在Vero E6细胞中显著抑制SARS-CoV的侵入和复制(EC50 = 300 mg·L-1, CC50 > 20 000 mg·L-1), 是具有开发前景的先导化合物。早期对甘草酸的构效关系的研究表明, 甘草酸碳水化合物链的延长或氨基酸/杂环片段的引入对糖苷的生物活性有显著的影响[58]。基于此, 2005年Hoever等[59]以甘草酸为先导化合物, 在细胞活性水平上(Vero E6细胞)测试了15个甘草酸衍生物对SARS-CoV的抑制活性。结果表明在甘草酸的糖苷链中引入2-乙酰氨基-D-葡萄糖所得的化合物8b, 活性最优, 比先导化合物提高了10倍左右(EC50 = 40 ± 13 μmol·L-1, CC50 > 3 000 μmol·L-1, SI > 75), 但作用机制尚不清楚。此外研究结果还表明甘草酸的3位糖基片段和11位的羧基对抑制SARS病毒的活性至关重要, 这为后续的结构优化提供了指导。2020年2月3日, 斯坦福大学与香港大学研究团队[60]证明甘草酸可以与SARS-CoV-2的受体ACE2结合, 表明甘草酸制剂对SARS-CoV-2感染具有潜在的应用价值。此外, 有研究表明SARS-CoV-2的S蛋白上的RBD区域与SARS-CoV的RBD区域结构具有73.5%的相似性。经分子重叠和对接分析发现虽然与SARS相比, SARS-CoV-2的S蛋白RBD中的4个用于与宿主细胞ACE2蛋白结合的关键氨基酸发生了改变, 但由于RBD结构域的三维结构变化较小, 与ACE2蛋白仍然具备很强的结合自由能(-50.6 kcal·mol-1)。这也提示针对宿主受体ACE2进行新药研发可能会产生较理想的广谱抗冠状病毒药物[61]。
|
综上可得, 对SARS-CoV-2有效的老药可分为RNA聚合酶抑制剂、蛋白酶抑制剂、宿主受体ACE2抑制剂、溶酶体酸化以及干扰素反应(表 2), 这些药物的作用靶点在新冠病毒中都有类似的作用机制。可见, 在未来的研究中, 应把RNA病毒的共性作用机制或靶点作为重点以开发广谱抗病毒药物, 扩大可筛选的化合物库, 为新病毒的出现提供更多可能具有活性的候选药物。
| Table 2 Summary of old drugs that are effective for SARS-CoV-2 |
SARS-CoV3CL蛋白酶(SARS-CoV3CLpro)作为SARS-CoV主要的蛋白水解酶可切割SARS-CoV的两个多聚蛋白, 在病毒的生命周期中发挥了重要的作用, 成为SARS抑制剂研究的主要靶标。SARS-CoV3CLpro与HIV、HCV、登革病毒、肠病毒、西尼罗病毒的蛋白酶的三维结构具有一些共同的特征, 利用这些共同特征可以根据已知优势结构设计冠状病毒抑制剂[62-65]。
2005年, 台湾地区的研究者[66]基于人鼻病毒3C蛋白酶与SARS-CoV的3CLpro活性位点的相似性, 对靛红类人鼻病毒3C蛋白酶共价抑制剂进行体外测试, 发现对SARS-CoV 3CLpro具有较好的抑制活性(图 4), 其中化合物9活性最好(IC50 = 0.95 μmol·L-1)。2006年, 北京大学来鲁华课题组[67]基于靛红衍生物与SARS-CoV 3CLpro的结合方式, 为增加与靶标上His163、Phe140等关键残基的结合, 在结构骨架吲哚-2, 3-二酮的苯环上引入不同的氢键供受体基团, 获得活性提高的化合物10 (IC50 = 0.37 μmol·L-1)。进一步利用MALDI-TOFF质谱仪检测发现, 10与SARS-CoV 3CLpro可形成可逆非共价键, 这与靛红类衍生物与人鼻病毒3C蛋白酶形成的共价作用方式不同, 值得进一步研究。

|
Figure 4 The discovery of compounds 9-10 and binding mode of 10 with SARS-CoV 3CLpro. The hydrogen bonds are represented by red dotted lines |
α-酮酰胺片段具有两个氨基酸氢键受体, 常与催化位点的丝氨酸或半胱氨酸残基形成共价结合, 并可延长骨架、增加稳定性和透膜性, 是病毒蛋白酶抑制剂常见的优势片段。例如, 在丙肝病毒NS3/4A蛋白酶抑制剂特拉匹韦(11, telaprevir)的研究过程中, 用酮酰胺片段取代醛基明显增强了与靶标的结合(Ki值提高了约10倍), 并增加了稳定性[68, 69]。冠状病毒的主蛋白酶和肠病毒的3C蛋白酶具有相似的活性位点结构, 对底物P1位的谷氨酰胺有独特的要求, 并且在多种病毒的复制周期中都发挥着重要作用。这使得3C蛋白酶成为设计广谱抗病毒药物的理想靶点[70]。近期, Rolf Hilgenfeld课题组与中国科学院上海药物研究所在广谱抗冠状病毒领域取得了重要进展, 他们[71]根据含有α-酮酰胺基团的拟肽12与SARS-CoV Mpro等多种冠状病毒主蛋白酶共结晶分析, 发现α-酮酰胺可以和活性位点处的半胱氨酸形成共价相互作用。进而以该分子为先导对P1'、P2和P3位置进行结构优化, 发现了一类广谱抗病毒分子。其中, 化合物13和14在细胞水平的抗病毒活性实验中对MERS-CoV的EC50分别为0.4 nmol·L-1和7 nmol·L-1), 并且对肠病毒、α冠状病毒和β冠状病毒均具有微摩尔级别的抑制作用(图 5), 值得进一步深入研究以开发广谱抗病毒的候选药物。

|
Figure 5 Design of compounds 13 and 14 |
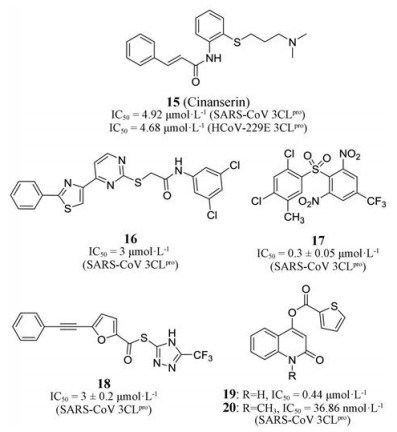
2005年, 中国科学院上海药物研究所的科学家[72]采用蛋白同源模建的方法利用基于靶标(SARS-CoV 3CLpro)的虚拟筛选对MDL公司的化合物库(MDL-CMC化合物库, 约8 000个分子)进行预测, 发现肉桂硫胺(15, cinanserin)与靶标结合能力最强, 并进一步通过表面等离子体共振技术证明了肉桂硫胺作用于SARS-CoV和HCoV-229E的3CLpro, 其KD = 49.4 μmol·L-1。抑酶活性测定发现肉桂硫胺对SARS-CoV 3CLpro和HCoV-229E的3CLpro的IC50值分别为4.92和4.68 μmol·L-1, 还具有较大的优化空间。这一研究也是老药新用又一成功案例。但需要注意的是, 肉桂硫胺是已知的5-羟色胺拮抗剂, 长期给药会有一定的不良反应。
2006年, Tsai等[73]基于SARS-CoV 3CLpro的晶体结构(PDB代码: 1UK4)对Maybridge数据库(Maybridge database, 约60 000个可用小分子)进行虚拟初筛, 获得了28个抑制剂(IC50 = 3~1 000 μmol·L-1), 其中化合物16活性最好, 对SARS-CoV 3CLpro的IC50值为3 μmol·L-1。此外, 研究人员进一步对这28个活性小分子进行3D-QSAR研究, 分析了靶标的结合位点以及配体的药效团, 为后期的优化提供了依据。
2006年, Lu等[74]利用GOLD version 2.1对接软件, 以SARS-CoV 3CLpro的三维结构(PDB代码: 1UK4)为基础, 对Maybridge数据库(约60 000个化合物)进行虚拟筛选, 获得21个具有抑制活性的小分子(IC50 < 50 μmol·L-1), 其中化合物17和18的抑制活性最好, IC50分别为0.3 ± 0.05和3 ± 0.2 μmol·L-1, 并通过分子模拟解释了构效关系。
此外, 南开大学团队利用Glide分子对接和Discovery Studio分子模拟软件生成基于受体结构的药效团模型对ZINC、InterBioScreening、TCM Database等多个数据库(60万小分子)进行虚拟筛选, 发现苗头化合物, 并经结构优化获得了对SARS-CoV 3CLpro具有较强抑制活性的19和20, IC50分别达到0.44 μmol·L-1和36.86 nmol·L-1 [75, 76]。
总之, 虚拟筛选在进行生物活性筛选之前, 利用分子对接软件模拟药物靶标与化合物之间的相互作用, 计算二者之间的亲和力大小, 以降低实际活性筛选的化合物数目, 有望显著扩大有效分子搜索的范围和精确度, 提高先导化合物发现效率。随着近几年人工智能与计算化学技术的发展, 虚拟筛选在此次新冠病毒疫情的药物筛选工作中也起到了重要作用。
2.3 基于病毒蛋白结构的生物活性筛选以分子水平的实验方法为基础, 通过数量庞大的药品样品库来对不同病毒靶标进行筛选, 可以快速寻找到优质先导化合物, 用于新药的研发。2005年, Kaeppler等[77]根据SARS-CoV 3CLpro的结构, 利用HPLC技术对含有亲电基团(氮杂环氧化物、迈克尔体系)的化合物进行筛选(这些含有亲电基团的化合物可以和SARS-CoV 3CLpro活性部位的亲核氨基酸反应)发现非肽类共价抑制剂21。21在100 μmol·L-1浓度下对SARS-CoV 3CLpro的抑制率达75%, Ki值为45.8 μmol·L-1。分子对接表明21横跨结合口袋(图 6), 叔丁基位于疏水的S4口袋, 二氯苯基位于靠近S3的位置, 迈克尔体系的末端乙基指向远离活性中心的位置, 并且整个分子结构可以和His163、Glu166和Gln189之间可以形成氢键作用。研究人员进一步探究发现在S4口袋处引入新的氢键供受体基团可能会增加与靶标的亲和力。随后对S4口袋处的叔丁酰胺基团进行构效关系研究, 发现用异丁酰胺替代化合物21的叔丁酰胺上的叔丁基所得的化合物22, 显著增加了与靶标的氢键作用, 活性略有提升(Ki = 35.5 μmol·L-1), 但仍具有较大的优化空间。
|

|
Figure 6 The structures and binding mode of 21 and 22 with SARS-CoV 3CLpro. The hydrogen bonds are represented by red dotted lines |
中国科学院上海药物研究所研究人员结合虚拟筛选、表面等离子共振技术(SPR)、荧光共振能量转移(FRET)等生物物理技术, 建立了一个高效的SARS-CoV 3CLpro抑制剂筛选平台[78]。从MDL-ACD化合物库中, 该团队筛选到了天然产物类SARS-CoV3CLpro抑制剂五羟黄酮-3-β-半乳糖苷(23, quercetin-3-galactoside), IC50值为42.8 μmol·L-1, KD值为38.4 μmol·L-1。通过分子模拟、点突变实验、FRET和SPR技术证明SARS-CoV3CLpro的Gln189残基对五羟黄酮-3-β-3半乳糖苷与酶的结合起重要作用。这一研究再次提示人们, 天然产物具有结构的多样性和生物活性的多样性等特点, 可作为高通量筛选的最具潜力的资源之一[79]。

|
2010年, 美国普渡大学的Ghosh等[80]以SARS PLpro为筛选模型, 对50 080个类药小分子进行高通量筛选, 获得活性化合物24 (6577871), IC50为59 μmol·L-1。随后, 研究人员解析了24与靶标PLpro的X-ray晶体结构(PDB代码: 3E9S), 指导该先导化合物精确的结构优化(图 7), 获得(S)-甲基化的抑制剂25 (IC50 = 0.56 ± 0.03 μmol·L-1)和(R)-甲基化的抑制剂26 (IC50 = 0.32 ± 0.01 μmol·L-1)。并测试了细胞(Vero E6)水平的抗病毒活性(EC50均为9.1 μmol·L-1)。2014年, Báez-Santos等[81]基于该类骨架, 经结构优化得到化合物27, 抑制活性大大提高(IC50 = 0.15 μmol·L-1), 但代谢不稳定, 化合物28和29对SARS-CoV PLpro的IC50分别为0.39和0.35 μmol·L-1, 在Vero细胞中抑制SARS-CoV感染的EC50分别为8.3和9.5 μmol·L-1, 且类药性较好, 可进一步进行体内试验研究。

|
Figure 7 The structures of compounds 24-29 |
2013年, Jacobs等[82, 83]对NIH分子库(约293 000个化合物)进行高通量筛选, 获得了101个对SARS-CoV 3CLpro具有抑制活性的化合物(IC50 < 10 μmol·L-1)。其中, 二肽化合物30 (SID49730186)具有较好的抑制活性与选择性(对3CLpro的IC50值为2.2 μmol·L-1, 对PLpro的IC50值为34 μmol·L-1, 图 8)。研究人员进而以其为先导化合物, 利用Ugi四组分反应快速高效地得到了一系列新衍生物, 并经构效关系研究, 获得具有较高活性和选择性的化合物31 (ML188), 抑制3CLpro的IC50值为1.5 μmol·L-1, 对PLpro无抑制活性。X-射线共晶结构(PDB代码: 3V3M)显示化合物31上的3-吡啶氮在S1口袋内作为氢键受体与His163结合, 呋喃和酰胺羰基上的氧原子与Gly143的主链NH具有双重氢键作用, 为进一步结构优化提供了信息。这也提示我们要充分利用组合化学、点击化学、多样性导向合成、微量合成技术等化合物库的高效合成技术, 并结合快速筛选系统, 对抑制剂进行系统的构效关系研究。

|
Figure 8 Structures of compounds 30, 31 (the key H-bond interactions between 31 and the target were illustrated) |
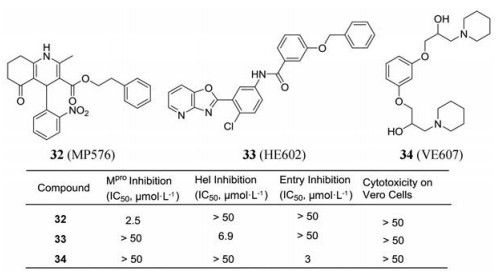
Kao等[84]通过高通量筛选和基于细胞水平(Vero E6)的表型筛选, 从ChemBridge Corporation化合物库(50 240个结构多样的小分子)中发现104种具有抑制SARS-CoV感染的化合物, 大多数的EC50在低微摩尔范围内。经化学基因组学研究发现, 2个为SARS-CoV 3CLpro抑制剂、7个为解旋酶(Hel)抑制剂、18种靶向S蛋白-ACE2介导的病毒进入以及剩余的78个化合物的作用靶点尚未确定。在这些靶点确定的化合物中活性最好的(EC50 < 10 μmol·L-1)为SARS-CoV 3CLpro抑制剂32 (MP576), Hel抑制剂33 (HE602)和病毒进入的抑制剂34 (VE607), 为治疗SARS-CoV病毒感染提供了结构新颖的可优化化合物。在本研究中, Kao等[85]将筛选技术和化学基因组学相结合, 高效地验证化合物在细胞内所干扰的致病途径, 是一种新型有效的方法, 值得进一步推广。
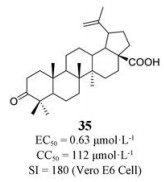
2007年, Wen等[86]测试了221种天然产物(从一些药用植物中分离出具有几个特定化学骨架的化合物)对SARS-CoV诱导的Vero E6细胞病变效应的影响, 从中筛选出20种活性较好的化合物(包括木脂素、姜黄素、松烷型和棕榈烷型二萜等), 其中化合物35活性最好(EC50 = 0.63 μmol·L-1, CC50 = 112 μmol·L-1)。这些天然产物活性成分首次被发现具有显著的抗SARS-CoV活性, 值得进一步研究。
|
|
2019年, 清华大学饶燏课题组[87]在细胞(HCV感染的Huh7.5.1细胞)水平上对课题组自主构建的微型化合物库(约200个喹诺酮类小分子)进行表型筛选, 获得活性较优的苗头化合物36 (RYL-552S) (EC50 = 0.36 μmol·L-1, CC50 > 10 μmol·L-1)。构效关系分析得先导化合物37 (RYL-634) (EC50 = 0.006 ± 0.007 μmol·L-1, CC50 > 2.5 μmol·L-1)。并且, 37对肠道病毒71 (EV71)、HIV、MERS-CoV和流感病毒等都具有广谱抑制活性, 且毒性较低(图 9)。进一步通过ABPP、反向对接等方法发现RYL-634的作用靶标为二氢乳酸脱氢酶(DHODH) (PDB代码: 4IGH)。其对接模式表明喹诺酮上的酮羰基和氟原子以及右侧苯环上的氮原子为氢键受体, 右侧的苯环可与靶标可形成π-π堆积作用, 这和构效关系分析结果基本吻合(图 9A)。喹诺酮是药物化学中的优势结构, 在抗病毒药物领域具有进一步研究的前景。

|
Figure 9 The structures of compounds 36-37 and binding model of RYL-634 with DHODH |
根据靶标结构和基于片段的高通量晶体筛选技术联合运用可以高效、快速地发现全新的苗头化合物, 经结构优化发现结构新颖的分子, 可作为发现抗冠状病毒药物的长期策略。SARS-CoV3CLpro具有明确的三维结构, 是运用高通量晶体筛选的理想对象[88-90]。南开大学团队[91, 92]运用蛋白质高通量结晶技术, 对小分子片段库进行共结晶筛选, 得到了SARS-CoV3CLpro与小分子5-溴靛红的共结晶复合物, 结合前人相关研究[66], 运用计算机辅助药物设计的方法, 发现了一系列靛红及喹啉类衍生物, 其中化合物1-活性萘甲基-5-溴靛红(38)和1-萘甲基-6-溴喹啉-2-酮(39)活性最好, IC50值分别为0.15和0.28 μmol·L-1, 另外5-磺胺类靛红(40a、40b和40c)抑制SARS-CoV3CLpro的IC50值均处于微摩尔水平。该研究和前期研究人员[66]运用基于靶标结构相似性的优势结构再定位策略所发现的靛红衍生的结构骨架类似, 但发现的途径截然不同, 再次验证了靛红类结构骨架的重要性。

|
作为组合化学中的新兴分支, 动态组合化学是一种集合成与筛选为一体的组合化学研究新方法。在动态组合化合物库中, 通过靶标分子的诱导结合驱动作用, 能够选择性地筛选到与靶标分子存在强相互作用的优势化合物(即动态偶联筛选, dynamic ligation screening, DLS), 因而动态组合化学往往与基于片段的药物设计相结合(图 10), 在药物研发领域有着广泛的应用前景[93]。基于该策略, Rademann J团队运用动态可逆的腙化学得到了化合物41, 抑制SARS-CoV-3CLpro的Ki值(HPLC方法)为2.9 μmol·L-1[94, 95]。

|
Figure 10 The concept of dynamic ligation screening (DLS) and the structure of compound 41. Substrate competes with peptide aldehyde (pink) for SARS-CoV-3CLpro (sky blue). Active fragment (blue) leads to an increased inhibition through the binding of the imine ligation product to the active site |
MERS-CoV属于I类融合包膜病毒。该类病毒利用其包膜上的I类融合蛋白作为介导分子, 与宿主细胞膜发生融合, 是该类病毒感染的关键环节。在该步骤中, 融合蛋白跨膜亚基会发生分子内折叠, 其C端重复序列(CHR)和N端重复序列(NHR)相互作用, 形成六股α螺旋束(6-HB)。基于病毒NHR或CHR片段的类肽结构, 可以通过干扰6-HB的形成, 进而抑制病毒与宿主细胞的融合, “御敌于国门之外”, 并被身体的免疫系统清除, 达到治疗的目的。I类融合包膜病毒在使用6-HB以驱动病毒和宿主细胞膜融合方面具有相似之处, 可据此设计广谱抗病毒多肽。2018年, 刘克良与姜世勃联合团队报道了靶向细胞融合的α-螺旋肽42, 对MERS-CoV (EC50 = 0.11 μmol·L-1, CC50 > 100 μmol·L-1)和甲型流感病毒(H1N1: EC50 = 1.73 μmol·L-1, CC50 > 100 μmol·L-1)具有显著抑制活性[96]。
同年, 该联合团队[97]基于MERS-CoVCHR区域的结构生物学信息(PDB代码: 4NJL), 针对融合蛋白上的CHR和NHR之间的起关键作用的残基, 设计了模拟MERS-CoV的S蛋白C末端短α-螺旋区的天然构象的全烃化装订多肽。其中43 (P21S10, LDLTYEMLSL QQVVK*LNE*Y)不仅保留了MERS-CoV融合机制中具有生物活性的α-螺旋构象, 而且具有较强的天然配体结合能力、良好的抗病毒活性(EC50 = 0.97 μmol·L-1, CC50 > 100 μmol·L-1, Huh-7 cells)和药代动力学性质。
在此次疫情中, 该团队[98]基于前期的研究, 对SARS-CoV和SARS-CoV-2的S蛋白的氨基酸序列进行比对, 发现S蛋白上的S2亚基高度保守, 在HR1和HR2结构域的总体同源性为96.2%和100%。在HR1核心区内, 21个氨基酸中有8个残基发生突变(相差约38%)。根据以往的经验该团队设计了HR1和HR2的衍生肽, 并利用凝胶电泳、圆二色谱法对其生物学特征进行研究, 发现SARS-CoV-2 S蛋白的HR1和HR2结构域可以相互作用形成6HB。进一步通过细胞-细胞融合实验对HR1和HR2的衍生肽以及以往设计的针对冠状病毒HR1域的融合抑制剂EK1进行研究, 发现HR2的衍生肽和EK1均可在低微摩尔浓度下抑制病毒的融合。这些研究表明SARS-CoV-2 S蛋白的HR1和HR2在介导SARS-CoV-2与宿主细胞融合以及宿主细胞的侵入中有着关键的作用, 并且具有与SARS-CoV相似的膜融合机制, 是小分子、多肽药物开发的潜在靶点。
2.8 基于宿主的药物分子设计ACE2蛋白是SARS冠状病毒的宿主受体, 在SARS-CoV侵入过程中可识别S蛋白并介导病毒与宿主细胞的结合和随后的进入, 是治疗干预的潜在靶标, 但鉴于该蛋白在心血管功能中的正常生理作用, 因此存在潜在的毒性[99-101]。2002年, Dales等[102]基于对ACE2羧肽酶的功能, 检测了其切割130种已知肽激素的碳末端残基的能力, 并通过底物筛选数据以及肽序列的比对经合理设计得到化合物44。该化合物具有碳末端羧酸的非水解性His-Leu类分子, 并在中心位置含有羧酸盐作为锌配位元件, 似乎可同时占据S1和S1', 对ACE2具有微摩尔浓度的抑制活性(图 11)。研究人员进一步进行构效关系研究发现, 具有纳摩尔级的抑制剂45 (MLN-4760, 图 11)对ACE2的IC50为0.44 nmol·L-1, 选择性较好。机制研究表明当MLN-4760与ACE2结合后, 两个ACE2亚结构域(可能是S1识别区域)的外边缘发生了巨大的构象变化, 这可能会破坏S蛋白和ACE2之间的相互作用, 从而干扰病毒的进入。值得一提的是该方法避免了传统的高通量筛选, 根据蛋白结构和底物的特点, 结合合理设计和结构优化成功获得具有纳摩尔级别的化合物, 值得参考。

|
Figure 11 Design of ACE2 inhibitor 45 |
近日, 西湖大学利用冷冻电镜技术成功解析SARS-CoV-2的受体—ACE2的全长结构[103]。研究发现ACE2以二聚体形式存在, 同时具有开放和关闭两种构象变化, 并且两种构象均含有与冠状病毒的相互识别界面。该发现有助于分析冠状病毒进入靶细胞的结构基础和功能特征, 对发现和优化阻断进入细胞的抑制剂有重要作用。
亲环素A (cyclophilin A, CypA)是亲环素家族重要成员, 是重要的免疫抑制药物环孢素A的体内结合蛋白, 除了具有肽脯氨酰顺反异构酶活性, 亲环素A在一系列生物途径中发挥作用, 如蛋白折叠、装配、转运、神经生长调节和病毒复制。2004年, 中国科学院上海药物研究所[104]发现SARS冠状病毒的核衣壳蛋白与人CypA具有较强的结合能力(KD = 6~160 nmol·L-1), 并且这种结合是功能性的, 这为治疗SARS-CoV感染提供了新线索[105]。
2009年, 华东理工大学、北京大学和上海药物研究所[106]合作利用基于靶标的全新药物设计策略结合结构优化、有机合成和生物测定, 发现了高效的CypA抑制剂。研发流程如图 12所示, 首先利用LigBuilder2.0程序结合前人的研究构建了CypA抑制剂的连接片段—酰脲(和靶标上的残基可形成氢键作用), 然后利用LigBuilder2.0程序根据CypA结合位点的形状和性质生成新的分子, 最终发现了一种高效的先导化合物46 (IC50 = 6.1 nmol·L-1)。进一步对先导46进行SAR研究得CypA抑制剂47和48 (IC50分别为2.59 ± 0.20、1.52 ± 0.10 nmol·L-1), 抑制活性明显提升, 是迄今为止报道的最有效的CypA抑制剂。相比于传统的虚拟筛选, 全新药物设计策略可以产生结构新颖的化合物, 不受已有结构的限制。但需要考虑合成可及性及分子的类药性。

|
Figure 12 The design idea of compounds 47 and 48 |
自疫情暴发以来, 相关部门即出台相应政策支持针对COVID-19的相关治疗药物研发。众所周知抗病毒药物的研发通常需要大量的资源和时间, 尽管目前具有药物研发的软硬件条件, 但在应对此次突发性的、高传染性的病原体所造成的公共卫生危机时, 常规的药物研发思路显然“远水不及近渴”[107], 正如有的学者所说“不管疾病有多严重, 不管我们期待新药和新疫苗的愿望是多么迫切, 新药和新疫苗开发的规律无法被逾越”[108]。药物研发有其固有的规律, “逆者难从, 顺着易行”, 绝非人的意志可以转移。
面对疫情, 老药新用的优势就是其安全性已经经过验证, 可以绕过动物安全性研究(PK/PD和动物毒性实验), 直接进入临床Ⅱ或Ⅲ期, 并且能够保证供应。可用的“老药”库包括已上市药物库、已证明安全性的临床期化合物库或广谱抗病毒化合物库(包括靶向宿主的药物, 如阿比多尔、硝唑尼特等), 在选择筛选的药物库时, 大都是基于相似性的“就近”考虑, 优先考虑已知的对SRAS-CoV和MERS-CoV广谱抗冠状病毒药物[23, 109]及靶向宿主[60, 110]的广谱抗病毒药物, 新近针对SARS-CoV-2的药物研发, 大都体现了这种思路。“度以往事, 验之来事, 参之平素, 可则绝之”, 因此, 深挖抗SRAS-CoV和MERS-CoV药物研究文献(专利挖掘明显不够), 总结抗SARS-CoV、MERS-CoV等冠状病毒药物的成功经验与药物设计策略[111, 112], 有望为打赢当前疫情防控阻击战提供有价值的资讯。
无疑, 在新疫情暴发时的应急药物研发, 药物筛选方法的建立是首要环节。病毒学家能否快速分离病毒是建立细胞水平活性评价方法的前提; 病毒分子生物学家能否快速完成病毒基因测序对结构生物学家完成蛋白表达、结构解析至关重要。这些工作均依赖于规范化的生物安全实验室, 这也正是我国多数抗病毒药物研究机构的“卡脖子”环节。因此, 迫切需要增设规范化的生物安全实验室与抗病毒筛选平台, 在疫情暴发时便于与药物研发单位联合攻关, 为应对今后可能会出现的冠状病毒感染突发疫情奠定基础。
与此同时, 面对严峻的疫情, 当生物安全实验室缺乏, 药物筛选条件受限, 或当结构生物学等基础研究滞后; 或者待测化合物过多, 人力成本过高时, 计算机辅助药物设计作为现代药物研发的常规方法, 可加快特效药物的研发速度[113]。例如, 通过对病毒蛋白的序列比对可以发现, SARS-CoV-2的某些关键蛋白和SRAS-CoV的序列同源性高达95%以上。因此, 研究者可通过CADD技术, 根据SARS-CoV-2与蛋白质晶体结构数据库中其他已知冠状病毒的序列类似性, 进行同源模建, 构建出合理的SARS-CoV-2的相应蛋白质的结构, 进而进行基于靶标的虚拟筛选, 在新近的抗SARS-CoV-2药物研发中也多有应用[32, 114-116]。相信人工智能和大数据技术能够大大加快了新药设计的速度, 节省新药创制的人力和物力[117]。
疫情过后, 建议政府加大对冠状病毒以及广谱抗病毒药物基础研究的投入。针对特异性病毒靶标及宿主因子, 综合运用结构生物学、生物信息大数据、计算机辅助药物设计技术及药物设计新理念(如基于片段的药物设计、蛋白质高通量结晶筛选、共价结合、以蛋白为微反应器的片段自组装策略、蛋白降解靶向嵌合体以及高品质化合物库的构建技术等)[16], 开展药物设计及化合物库筛选, 发现骨架新颖、结构多样的苗头化合物, 进而通过构效关系研究、结构优化及成药性评价, 发现先导化合物、候选药物及临床有效药物, 丰富抗病毒药物“储备库”, 以备抗“疫”之需(图 13)。

|
Figure 13 Schematic diagram of current and future anti-coronavirus drug research model |
| [1] |
Perlman S, Netland J. Coronaviruses post-SARS: update on replication and pathogenesis[J]. Nat Rev Microbiol, 2009, 7: 439-450. DOI:10.1038/nrmicro2147 |
| [2] |
Stadler K, Masignani V, Eickmann M, et al. SARS--beginning to understand a new virus[J]. Nat Rev Microbiol, 2003, 1: 209-218. DOI:10.1038/nrmicro775 |
| [3] |
Graham RL, Donaldson EF, Baric RS. A decade after SARS: strategies for controlling emerging coronaviruses[J]. Nat Rev Microbiol, 2013, 11: 836-848. |
| [4] |
de Wit E, van Doremalen N, Falzarano D, et al. SARS and MERS: recent insights into emerging coronaviruses[J]. Nat Rev Microbiol, 2016, 14: 523-534. DOI:10.1038/nrmicro.2016.81 |
| [5] |
Du Toit A. Outbreak of a novel coronavirus[J]. Nat Rev Microbiol, 2020. DOI:10.1038/s41579-020-0332-0 |
| [6] | |
| [7] |
Zumla A, Chan JF, Azhar EI, et al. Coronaviruses - drug discovery and therapeutic options[J]. Nat Rev Drug Discov, 2016, 15: 327-347. DOI:10.1038/nrd.2015.37 |
| [8] |
Jiang S, Du L, Shi Z. An emerging coronavirus causing pneumonia outbreak in Wuhan, China: calling for developing therapeutic and prophylactic strategies[J]. Emerg Microbes Infect, 2020, 9: 275-277. DOI:10.1080/22221751.2020.1723441 |
| [9] |
Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses[J]. Nat Rev Microbiol, 2019, 17: 181-192. DOI:10.1038/s41579-018-0118-9 |
| [10] |
Lomniczi B. Biological properties of avian coronavirus RNA[J]. J Gen Virol, 1977, 36: 531-533. DOI:10.1099/0022-1317-36-3-531 |
| [11] |
Chan JF, Lau SK, To KK, et al. Middle East respiratory syndrome coronavirus: another zoonotic betacoronavirus causing SARS-like disease[J]. Clin Microbiol Rev, 2015, 28: 465-522. |
| [12] |
Raj VS, Mou H, Smits SL, et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC[J]. Nature, 2013, 495: 251-254. DOI:10.1038/nature12005 |
| [13] |
Li W, Moore MJ, Vasilieva N. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus[J]. Nature, 2003, 426: 450-454. DOI:10.1038/nature02145 |
| [14] |
Du L, He Y, Zhou Y, et al. The spike protein of SARS-CoV--a target for vaccine and therapeutic development[J]. Nat Rev Microbiol, 2009, 7: 226-236. DOI:10.1038/nrmicro2090 |
| [15] |
Pillaiyar T, Manickam M, Namasivayam V, et al. An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy[J]. J Med Chem, 2016, 59: 6595-6628. DOI:10.1021/acs.jmedchem.5b01461 |
| [16] |
Wu GC, Zhao T, Kang DW, et al. Overview of recent strategic advances in medicinal chemistry[J]. J Med Chem, 2019, 62: 9375-9414. DOI:10.1021/acs.jmedchem.9b00359 |
| [17] |
Zhan P, Pannecouque C, De Clercq E, et al. Anti-HIV drug discovery and development: current innovations and future trends[J]. J Med Chem, 2016, 59: 2849-2878. DOI:10.1021/acs.jmedchem.5b00497 |
| [18] |
Zhan P, Liu X. A review of new strategies for the discovery and optimization of lead compounds (Part Ⅰ)[J]. China Sciencepaper(中国科技论文), 2015(24): 2918-2928. |
| [19] |
Delang L, Abdelnabi R, Neyts J, et al. Favipiravir as a potential countermeasure against neglected and emerging RNA viruses[J]. Antiviral Res, 2018, 153: 85-94. DOI:10.1016/j.antiviral.2018.03.003 |
| [20] |
Delang L, Segura GN, Tas A, et al. Mutations in the chikungunya virus non-structural proteins cause resistance to favipiravir (T-705), a broad-spectrum antiviral[J]. J Antimicrob Chemother, 2014, 69: 2770-2784. DOI:10.1093/jac/dku209 |
| [21] | |
| [22] | |
| [23] |
Wang ML, Cao RY, Zhang LK, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro[J]. Cell Research, 2020. DOI:10.1038/s41422-020-0282-0 |
| [24] | |
| [25] |
Holshue ML, Debolt C, Lindquist S, et al. First case of 2019 novel coronavirus in the United States[J]. N Engl J Med, 2020. DOI:10.1056/NEJMoa2001191 |
| [26] |
Guo ZR. Remdesivir phase Ⅲ clinical study developed from sympathetic medication[J]. Acta Pharm Sin (药学学报), 2020, 55: 345-348. |
| [27] | |
| [28] |
Ölschläger S, Neyts J, Günther S. Depletion of GTP pool is not the predominant mechanism by which ribavirin exerts its antiviral effect on Lassa virus[J]. Antiviral Res, 2011, 91: 89-93. DOI:10.1016/j.antiviral.2011.05.006 |
| [29] |
Ströher U, DiCaro A, Li Y, et al. Severe acute respiratory syndrome-related coronavirus is inhibited by interferon-a[J]. J Infect Dis, 2004, 189: 1164-1167. DOI:10.1086/382597 |
| [30] |
Chen F, Chan KH, Jiang Y, et al. In vitro susceptibility of 10 clinical isolates of SARS coronavirus to selected antiviral compounds[J]. J Clin Virol, 2004, 31: 69-75. |
| [31] |
Morgenstern B, Michaelis M, Baer PC, et al. Ribavirin and interferon-β synergistically inhibit SARS-associated coronavirus replication in animal and human cell lines[J]. Biochem Biophys Res Commun, 2005, 326: 905-908. DOI:10.1016/j.bbrc.2004.11.128 |
| [32] | |
| [33] |
Plantone D, Koudriavtseva T. Current and future use of chloroquine and hydroxychloroquine in infectious, immune, neoplastic, and neurological diseases: a mini-review[J]. Clin Drug Investig, 2018, 38: 653-671. DOI:10.1007/s40261-018-0656-y |
| [34] |
Keyaerts E, Li S, Vijgen L, et al. Antiviral activity of chloroquine against human coronavirus OC43 infection in newborn mice[J]. Antimicrob Agents Chemother, 2009, 53: 3416-3421. DOI:10.1128/AAC.01509-08 |
| [35] | |
| [36] |
Rolain JM, Colson P, Raoult D. Recycling of chloroquine and its hydroxyl analogue to face bacterial, fungal and viral infections in the 21st century[J]. Int J Antimicrob Agents, 2007, 30: 297-308. DOI:10.1016/j.ijantimicag.2007.05.015 |
| [37] |
Salata C, Calistri A, Parolin C, et al. Antiviral activity of cationic amphiphilic drugs[J]. Expert Rev Anti Infect Ther, 2017, 15: 483-492. DOI:10.1080/14787210.2017.1305888 |
| [38] | |
| [39] |
Cohen SA. Use of nitazoxanide as a new therapeutic option for persistent diarrhea: a pediatric perspective[J]. Curr Med Res Opin, 2005, 21: 999-1004. DOI:10.1185/030079905X50534 |
| [40] |
Fox LM, Saravolatz LD. A new thiazolide antiparasitic agent[J]. Clin Infect Dis, 2005, 40: 1173-1180. DOI:10.1086/428839 |
| [41] |
Rossignol JF. Nitazoxanide: a first-in-class broad-spectrum antiviral agent[J]. Antiviral Res, 2014, 110: 94-103. DOI:10.1016/j.antiviral.2014.07.014 |
| [42] |
Rossignol JF, La Frazia S, Chiappa L, et al. Thiazolides, a new class of anti-influenza molecules targeting viral hemagglutinin at the post-translational level[J]. J Biol Chem, 2009, 284: 29798-29808. DOI:10.1074/jbc.M109.029470 |
| [43] |
Cao J, Forrest JC, Zhang X. A screen of the NIH Clinical Collection small molecule library identifies potential anti-coronavirus drugs[J]. Antiviral Res, 2015, 114: 1-10. DOI:10.1016/j.antiviral.2014.11.010 |
| [44] |
Rossignol JF. Nitazoxanide, a new drug candidate for the treatment of Middle East respiratory syndrome coronavirus[J]. J Infect Public Health, 2016, 9: 227-230. DOI:10.1016/j.jiph.2016.04.001 |
| [45] | |
| [46] |
Wu CJ, Jan JT, Chen CM, et al. Inhibition of severe acute respiratory syndrome coronavirus replication by niclosamide[J]. Antimicrob Agents Chemother, 2004, 48: 2693-2696. DOI:10.1128/AAC.48.7.2693-2696.2004 |
| [47] | |
| [48] |
Sheahan TP, Sims AC, Leist SR, et al. Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV[J]. Nat Commun, 2020, 11: 222. DOI:10.1038/s41467-019-13940-6 |
| [49] |
Arabi YM, Asiri AY, Assiri AM, et al. Treatment of Middle East respiratory syndrome with a combination of lopinavir/ritonavir and interferon-β1b (MIRACLE trial): statistical analysis plan for a recursive two-stage group sequential randomized controlled trial[J]. Trials, 2020, 21: 8. DOI:10.1186/s13063-019-3846-x |
| [50] |
Arabi YM, Alothman A, Balkhy HH, et al. Treatment of Middle East respiratory syndrome with a combination of lopinavir-ritonavir and interferon-β1b (MIRACLE trial): study protocol for a randomized controlled trial[J]. Trials, 2018, 19: 81. DOI:10.1186/s13063-017-2427-0 |
| [51] |
Chu CM, Cheng VC, Hung IF, et al. Role of lopinavir/ritonavir in the treatment of SARS: initial virological and clinical findings[J]. Thorax, 2004, 59: 252-256. DOI:10.1136/thorax.2003.012658 |
| [52] | |
| [53] |
Cheng VC, Chan JF, To KK, et al. Clinical management and infection control of SARS: lessons learned[J]. Antiviral Res, 2013, 100: 407-419. DOI:10.1016/j.antiviral.2013.08.016 |
| [54] |
Cinatl J Jr, Michaelis M, Scholz M, et al. Role of interferons in the treatment of severe acute respiratory syndrome[J]. Expert Opin Biol Ther, 2004, 4: 827-836. DOI:10.1517/14712598.4.6.827 |
| [55] |
Falzarano D, de Wit E, Martellaro C, et al. Inhibition of novel beta coronavirus replication by a combination of interferon-alpha 2b and ribavirin[J]. Sci Rep, 2013, 3: 1686. DOI:10.1038/srep01686 |
| [56] |
Sun ZG, Zhao TT, Lu N, et al. Research progress of glycyrrhizic acid on antiviral activity[J]. Mini Rev Med Chem, 2019, 19: 826-832. DOI:10.2174/1389557519666190119111125 |
| [57] |
Cinatl JMB, Bauer G, Chandra P, et al. Glycyrrhizin, an active component of liquorice roots, and replication of SARS-associated coronavirus[J]. Lancet, 2003, 361: 2045-2046. DOI:10.1016/S0140-6736(03)13615-X |
| [58] |
Wu CY, Jan JT, Ma SH, et al. Small molecules targeting severe acute respiratory syndrome human coronavirus[J]. Proc Natl Acad Sci U S A, 2004, 101: 10012-10017. DOI:10.1073/pnas.0403596101 |
| [59] |
Hoever G, Baltina L, Michaelis M, et al. Antiviral activity of glycyrrhizic acid derivatives against SARS-Coronavirus[J]. J Med Chem, 2005, 48: 1256-1259. DOI:10.1021/jm0493008 |
| [60] | |
| [61] |
Xu XT, Chen P, Wang JF, et al. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission[J]. Sci China Life Sci, 2020. DOI:10.1007/s11427-020-1637-5 |
| [62] |
Steuber H, Hilgenfeld R. Recent advances in targeting viral proteases for the discovery of novel antivirals[J]. Curr Top Med Chem, 2010, 10: 323-345. DOI:10.2174/156802610790725470 |
| [63] |
Anand K, Ziebuhr J, Wadhwani P, et al. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs[J]. Science, 2003, 300: 1763-1767. DOI:10.1126/science.1085658 |
| [64] |
Kaiser L, Crump CE, Hayden FG. In vitro activity of pleconaril and AG7088 against selected serotypes and clinical isolates of human rhinoviruses[J]. Antiviral Res, 2000, 47: 215-220. DOI:10.1016/S0166-3542(00)00106-6 |
| [65] |
Chou KC, Wei DQ, Zhong WZ, et al. Binding mechanism of coronavirus main proteinase with ligands and its implication to drug design against SARS[J]. Biochem Biophys Res Commun, 2003, 308: 148-151. DOI:10.1016/S0006-291X(03)01342-1 |
| [66] |
Chen LR, Wang YC, Lin YW, et al. Synthesis and evaluation of isatin derivatives as effective SARS coronavirus 3CL protease inhibitors[J]. Bioorg Med Chem Lett, 2005, 15: 3058-3062. DOI:10.1016/j.bmcl.2005.04.027 |
| [67] |
Zhou L, Liu Y, Zhang W, et al. Isatin compounds as noncovalent SARS coronavirus 3C-like protease inhibitors[J]. J Med Chem, 2006, 49: 3440-3443. DOI:10.1021/jm0602357 |
| [68] |
Perni RB, Britt SD, Court JC, et al. Inhibitors of hepatitis C virus NS3/4A protease 1. Non-charged tetrapeptide variants[J]. Bioorg Med Chem Lett, 2003, 13: 4059-4063. DOI:10.1016/j.bmcl.2003.08.050 |
| [69] |
Perni RB, Farmer LJ, Cottrell KM, et al. Inhibitors of hepatitis C virus NS3/4A protease. Part 3: P2 proline variants[J]. Bioorg Med Chem Lett, 2004, 14: 1939-1942. DOI:10.1016/j.bmcl.2004.01.078 |
| [70] |
Anand K, Palm GJ, Mesters JR, et al. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain[J]. EMBO J, 2002, 21: 3213-3224. DOI:10.1093/emboj/cdf327 |
| [71] |
Zhang L, Lin D, Kusov Y, et al. Alpha-ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication Structure-based design, synthesis, and activity assessment[J]. J Med Chem, 2020. DOI:10.1021/acs.jmedchem.9b01828 |
| [72] |
Chen L, Gui C, Luo X, et al. Cinanserin is an inhibitor of the 3C-like proteinase of severe acute respiratory syndrome coronavirus and strongly reduces virus replication in vitro[J]. J Virol, 2005, 79: 7095-7103. DOI:10.1128/JVI.79.11.7095-7103.2005 |
| [73] |
Tsai KC, Chen SY, Liang PH, et al. Discovery of a novel family of SARS-CoV protease inhibitors by virtual screening and 3D-QSAR Studies[J]. J Med Chem, 2006, 49: 3485-3495. DOI:10.1021/jm050852f |
| [74] |
Lu IL, Mahindroo N, Liang PH, et al. Structure-based drug design and structural biology study of novel nonpeptide inhibitors of severe acute respiratory syndrome coronavirus main protease[J]. J Med Chem, 2006, 49: 5154-5161. DOI:10.1021/jm060207o |
| [75] |
Wang J. Structure-Based Drug Design of Novel Nonpeptide Inhibitors of Severe Acute Respiratory Syndrome Coronavirus Main Protease (基于结构针对SARS冠状病毒主蛋白酶的非肽类抑制剂设计) [D]. Tianjin: Nankai University, 2013.
|
| [76] |
Sun YP. Reasch of Quinolinone Compounds as SARS-CoV 3CLpro Inhibitors (喹啉酮类SARS-CoV 3CL蛋白酶抑制剂研究) [D]. Tianjin: Nankai University, 2013.
|
| [77] |
Kaeppler U, Stiefl N, Schiller M, et al. A new lead for nonpeptidic active-site-directed inhibitors of the severe acute respiratory syndrome coronavirus main protease discovered by a combination of screening and docking methods[J]. J Med Chem, 2005, 48: 6832-6842. DOI:10.1021/jm0501782 |
| [78] |
Chen LL. (1) The Discovery of 3CLpro Inhibitor of SARS Coronavirus, (2) The Study of Drug Absorption and Distribution Based on SPR Technique ((1) SARS冠状病毒3CL蛋白酶抑制剂的发现; (2)基于SPR技术的药物吸收和分布性质研究) [D]. Shanghai: Chinese Academy of Sciences Shanghai Institute of Materia Medica, 2006.
|
| [79] |
Wang P. Screening SARS Coronavirus Main Protease Inhibitors from Natural Products (天然产物中SARS冠状病毒主蛋白酶抑制剂的筛选) [D]. Tianjin: Nankai University, 2012.
|
| [80] |
Ghosh AK, Takayama J, Rao KV, et al. Severe acute respiratory syndrome coronavirus papain-like novel protease inhibitors: design, synthesis, protein-ligand X-ray structure and biological evaluation[J]. J Med Chem, 2010, 53: 4968-4979. DOI:10.1021/jm1004489 |
| [81] |
Báez-Santos YM, Barraza SJ, Wilson MW, et al. X-ray structural and biological evaluation of a series of potent and highly selective inhibitors of human coronavirus papain-like proteases[J]. J Med Chem, 2014, 57: 2393-2412. DOI:10.1021/jm401712t |
| [82] |
Jacobs J, Grum-Tokars V, Zhou Y, et al. Discovery, synthesis, and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease[J]. J Med Chem, 2013, 56: 534-546. DOI:10.1021/jm301580n |
| [83] |
Barretto N, Jukneliene D, Ratia K, et al. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity[J]. J Virol, 2005, 79: 15189-15198. DOI:10.1128/JVI.79.24.15189-15198.2005 |
| [84] |
Kao RY, Tsui WH, Lee TS, et al. Identification of novel small-molecule inhibitors of severe acute respiratory syndrome-associated coronavirus by chemical genetics[J]. Chem Biol, 2004, 11: 1293-1299. DOI:10.1016/j.chembiol.2004.07.013 |
| [85] |
Stockwell BR. Chemical genetics: ligand-based discovery of gene function[J]. Nat Rev Genet, 2000, 1: 116-125. DOI:10.1038/35038557 |
| [86] |
Wen CC, Kuo YH, Jan JT, et al. Specific plant terpenoids and lignoids possess potent antiviral activities against severe acute respiratory syndrome coronavirus[J]. J Med Chem, 2007, 50: 4087-4095. DOI:10.1021/jm070295s |
| [87] |
Yang Y, Cao L, Gao H, et al. Discovery, optimization, and target identification of novel potent broad-spectrum antiviral inhibitors[J]. J Med Chem, 2019, 62: 4056-4073. DOI:10.1021/acs.jmedchem.9b00091 |
| [88] |
Hilgenfeld R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design[J]. FEBS J, 2014, 281: 4085-4096. DOI:10.1111/febs.12936 |
| [89] |
Anand K, Ziebuhr J, Wadhwani P, et al. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs[J]. Science, 2003, 300: 1763-1767. DOI:10.1126/science.1085658 |
| [90] |
Verschueren KH, Pumpor K, Anemüller S, et al. A structural view of the inactivation of the SARS coronavirus main proteinase by benzotriazole esters[J]. Chem Biol, 2008, 15: 597-606. DOI:10.1016/j.chembiol.2008.04.011 |
| [91] |
Sun P. Design and Synthesis of the Inhibitor of SARS-CoV Mpro (SARS冠状病毒主蛋白酶抑制剂的设计与合成) [D]. Tianjin: Nankai University, 2012.
|
| [92] |
Zhu HM. Design, Synthesis and SAR Study of Isatin Derivatives as SARS CoV 3CLpro Inhibitors (靛红类SARS主蛋白酶(SARS CoV 3CLpro)抑制剂的设计、合成与构效关系研究) [D]. Tianjin: Nankai University, 2013.
|
| [93] |
He W, She PW, Fang Z, et al. The research progress of dynamic combinatorial chemistry[J]. Acta Pharm Sin (药学学报), 2013, 48: 814-823. |
| [94] |
Schmidt MF, Isidro-Llobet A, Lisurek M, et al. Sensitized detection of inhibitory fragments and iterative development of non-peptidic protease inhibitors by dynamic ligation screening[J]. Angew Chem Int Ed Engl, 2008, 47: 3275-3278. DOI:10.1002/anie.200704594 |
| [95] |
Tan J, Verschueren KH, Anand K, et al. pH-dependent conformational flexibility of the SARS-CoV main proteinase (M(pro)) dimer: molecular dynamics simulations and multiple X-ray structure analyses[J]. J Mol Biol, 2005, 354: 25-40. DOI:10.1016/j.jmb.2005.09.012 |
| [96] |
Wang C, Zhao L, Xia S, et al. De novo design of α-helical lipopeptides targeting viral fusion proteins: a promising strategy for relatively broad-spectrum antiviral drug discovery[J]. J Med Chem, 2018, 61: 8734-8745. DOI:10.1021/acs.jmedchem.8b00890 |
| [97] |
Wang C, Xia S, Zhang P, et al. Discovery of hydrocarbon-stapled short alpha-helical peptides as promising middle east respiratory syndrome coronavirus (MERS-CoV) fusion inhibitors[J]. J Med Chem, 2018, 61: 2018-2026. DOI:10.1021/acs.jmedchem.7b01732 |
| [98] |
Xia S, Zhu Y, Liu M, et al. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein[J]. Cell Mol Immunol, 2020. DOI:10.1038/s41423-020-0374-2 |
| [99] |
Song W, Gui M, Wang X, et al. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2[J]. PLoS Pathog, 2018, 14: e1007236. DOI:10.1371/journal.ppat.1007236 |
| [100] |
Struck AW, Axmann M, Pfefferle S, et al. A hexapeptide of the receptor-binding domain of SARS corona virus spike protein blocks viral entry into host cells via the human receptor ACE2[J]. Antiviral Res, 2012, 94: 288-296. DOI:10.1016/j.antiviral.2011.12.012 |
| [101] |
Yeung KS, Yamanaka GA, Meanwell NA. Severe acute respiratory syndrome coronavirus entry into host cells: opportunities for therapeutic intervention[J]. Med Res Rev, 2006, 26: 414-433. DOI:10.1002/med.20055 |
| [102] |
Dales NA, Gould AE, Brown JA, et al. Substrate-based design of the first class of angiotensin-converting enzyme-related carboxypeptidase (ACE2) inhibitors[J]. J Am Chem Soc, 2002, 124: 11852-11853. DOI:10.1021/ja0277226 |
| [103] |
Zhou Q, Yan RH, Zhang YY, et al. Structure of dimeric full-length human ACE2 in complex with B0AT1[J]. BioRxiv, 2020. DOI:10.1101/2020.02.17.951848 |
| [104] |
Luo C, Luo H, Zheng S, et al. Nucleocapsid protein of SARS coronavirus tightly binds to human cyclophilin A[J]. Biochem Biophys Res Commun, 2004, 321: 557-565. |
| [105] |
Pfefferle S, Schöpf J, Kögl M, et al. The SARS-coronavirus-host interactome: identification of cyclophilins as target for pan-coronavirus inhibitors[J]. PLoS Pathog, 2011, 7: e1002331. DOI:10.1371/journal.ppat.1002331 |
| [106] |
Ni S, Yuan Y, Huang J, et al. Discovering potent small molecule inhibitors of cyclophilin A using de novo drug design approach[J]. J Med Chem, 2009, 52: 5295-5298. DOI:10.1021/jm9008295 |
| [107] | |
| [108] | |
| [109] | |
| [110] |
Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin[J]. Nature, 2020. DOI:10.1038/s41586-020-2012-7 |
| [111] |
Liu W, Morse JS, Lalonde T, et al. Learning from the past: possible urgent prevention and treatment options for severe acute respiratory infections caused by 2019-nCoV[J]. Chembiochem, 2020. DOI:10.1002/cbic.202000047 |
| [112] |
Liu QY, Wang XL. Strategies for the development of drugs targeting novel coronavirus 2019-nCoV[J]. Acta Pharm Sin (药学学报), 2020. |
| [113] | |
| [114] | |
| [115] | |
| [116] |
Wu CR, Liu Y, Yang YY, et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods[J]. Acta Pharm Sin B, 2020. DOI:10.1016/j.apsb.2020.02.008 |
| [117] |
Richardson P, Griffin I, Tucker C, et al. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease[J]. Lancet, 2020. DOI:10.1016/S0140-6736(20)30304-4 |