 2020, Vol. 55
2020, Vol. 55



2. 北京大学药学院, 天然药物及仿生药物国家重点实验室, 北京 100191;
3. 中南大学湘雅医院皮肤科, 湖南 长沙 410012;
4. 云南中医医院制剂室, 云南 昆明 650021
2. State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, Beijing 100191, China;
3. The Department of Dermatology, Xiangya Hospital, Central South University, Changsha 410012, China;
4. Pharmaceutical Department, Yunnan Provincial Hospital of Traditional Chinese Medicine, Kunming 650021, China
蛋白水解靶向嵌合分子(protein proteolysis-targeting chimeras, PROTAC)是Crews等[1]于2001年首次报道的一种利用泛素-蛋白酶体降解蛋白的化合物。其由E3泛素连接酶配体和蛋白质配体, 通过连接基组合而成。E3泛素连接酶给目标靶蛋白加上泛素化蛋白标签, 可以启动细胞内强大的泛素化水解过程, 利用泛素-蛋白酶体途径特异性地降解靶蛋白。蛋白质降解可以通过靶蛋白表面上的任何结合位点介导, 而不是局限于单个可识别的活性位点, 可能更容易开发简单有效和选择性高配体, 使没有成药性的蛋白质具有成药价值; 同时, 降解蛋白不会出现抑制蛋白时的代偿性增加或突变情况, 可以有效解决耐药性问题, 所以降解靶蛋白的特性使PROTAC分子具有将无法成药蛋白质作为靶蛋白的潜力以及可能在药物耐药中发挥作用的优势[2]。最初的多肽类PROTAC分子存在着较大的跨膜问题与药效问题, 故现研究重点多在小分子类PROTAC上。现在的PROTAC分子设计的E3泛素连接酶配体部分多选用鼠双微基因(murine double minute 2, MDM2)、细胞凋亡抑制蛋白(cellular inhibitor of apoptosis protein, cIAP)、希佩尔-林道蛋白(von Hippel-Lindau protein, pVHL)和Cereblon (CRBN)的配体, 已有文献[3, 4]对4种E3连接酶的PROTAC小分子进行了相关报道, 但是PROTAC分子设计仍多为随机性, 具体的适用靶点仍未阐述清楚, 也未进行系统的总结。本文基于现有报道PROTAC小分子应用较多的靶蛋白, 根据其在细胞中的定位进行的分类, 整理出现阶段PTOTAC小分子设计选择靶点的倾向性。
1 以核内蛋白为靶点的应用细胞核内存在的蛋白质包括核受体、组蛋白等。核受体是后生动物中含量最丰富的转录调节因子之一, 它们在新陈代谢、性别决定与分化、生殖发育和稳态的维持等方面发挥着重要的功能。组蛋白是染色体基本结构蛋白, 它的修饰与基因表达调控有着密切的关系。核内蛋白在遗传及蛋白质合成里起着决定性作用, 多数疾病的发生发展也与核内蛋白的异常表达及调控密切相关。现已报道的PROTAC小分子多以核受体超家族及溴结构域和额外末端家族为靶点, 希望通过降解相应蛋白解决耐药性的问题。
1.1 雄激素受体(androgen receptor, AR)AR属于核受体超家族中的类固醇受体, 在体内起着重要的作用。除了与生殖相关外, 还具有保持体内激素平衡、刺激蛋白质合成代谢、促进氮沉积和增加肌纤维的数量和厚度等作用。雄激素受体拮抗剂广泛用于治疗前列腺癌(prostate cancer, PC)。这些拮抗剂通过抑制雄激素与AR的结合来抑制雄激素信号传导。目前, 临床上已在使用的雄激素受体拮抗剂如氟他胺、比卡鲁胺和恩杂鲁胺治疗PC。这种治疗虽然最初是有效的, 但是在长期使用雄激素受体拮抗剂后, 相当多的患者群体因为AR表达水平增加发展为去势抵抗性前列腺癌(castration-resistant prostate cancer, CRPC)。下调AR水平被认为是治疗CRPC的有效策略[5], 而PROTAC可以作为一种可以下调AR水平的选择。
小分子PROTAC的首次合成即为可降解体内AR的SARM-nutlin PROTAC[6]。之后, Shibata等[5]也合成了SNIPER (AR), 成功抑制了AR依赖性增殖的前列腺癌细胞的增殖活性; 王少萌等[7]合成了ARD-69, 能够在前列腺癌细胞系中将AR蛋白水平降低, 并有效抑制AR调节的基因表达。ARD-69抑制AR阳性前列腺癌细胞系中的细胞生长的效力比AR拮抗剂的效力高100倍, 为治疗CRPC开发药物提供了思路。
1.2 溴结构域和额外末端(bromodomain and extra-terminal, BET)蛋白家族含溴结构域蛋白(bromodomain-containing proteins)发挥表观遗传阅读器的功能。其中包含BRD2、BRD3、BRD4及BRDT的溴结构域和额外末端蛋白(BET)家族已成为肿瘤、炎症、HIV感染、中枢神经系统紊乱、心血管疾病的治疗靶点。所有BET家族成员均含有两个溴结构域(BD1和BD2), 通过结合组蛋白尾部的乙酰化赖氨酸残基调控基因转录[8]。
去势抵抗性前列腺癌的发生发展依赖于雄激素受体。其治疗靶点除了提到的AR之外, 溴结构域和末端外(BET)家族蛋白的抑制剂在CRPC的临床前模型中也显示出有效的活性。Raina等[9]合成的PROTAC分子ARV-771可以降解BET蛋白, 并且研究表明相比于BET抑制剂, ARV-771可以显著引起细胞凋亡。这验证了BET蛋白降解是一种治疗CRPC有前景的临床策略。
需要注意的是, BET家族中不同亚型也有其各自不同的功能, 如BRD4可替代靶向c-MYC, 作为急性髓性白血病的治疗靶点, 而其他亚型则无此功能。Zengerle等[10]开发了一种小分子PROTAC, 可用于靶向BET溴结构域并有效诱导BRD4的选择性降解。Lu等[11]用PROTAC分子ARV-825处理伯基特淋巴瘤(Burkitt's lymphoma, BL)细胞系, 发现此小分子导致BRD4蛋白质的几乎完全降解, 且与BRD4小分子抑制剂相比, 它可导致更显著和持久的c-MYC抑制。Winter等[12]所合成dBET1也证实了相比于抑制剂, 原代急性髓细胞白血病细胞对PROTAC分子的细胞凋亡反应有所增加。以上合成PROTAC分子, 均证明了与抑制剂相比, 靶蛋白降解剂的优势。
同时, 为了克服传统PROTAC分子细胞渗透、溶解度和其他类似药物的性质差的问题, Lebraud等[13]设计了可以通过两种较小前体的生物正交点击组合在细胞内形成的异双功能分子, 也成功地用于降解BRD4。
1.3 雌激素受体α (estrogen receptor α, ERα)ERα是核受体ER蛋白家族的关键成员, 控制着多种生理和发育过程。ERα通常在乳腺癌细胞中过表达并促进雌激素依赖性增殖, 这使其成为乳腺癌治疗的良好药物靶标。他莫昔芬与氟维司群都是可以竞争性结合ER并阻断ER促进的细胞增殖, 但大多数最初响应的乳腺肿瘤都具有抗药性。作为直接降解靶蛋白的策略, PROTAC技术可以潜在地解决这个问题。除PROTAC的多肽分子外, Ohoka等[14]合成了SNIPER (ER)小分子, 证明了SNIPER (ER)可诱导细胞中ERα蛋白的多泛素化, 并且它可以选择性地降解ERα而不会降解其他核受体。
1.4 周期蛋白依赖性激酶(cyclin-dependent kinases, CDK)CDK是与细胞周期进程相对应的一套Ser/Thr激酶系统。细胞周期蛋白依赖性激酶9 (CDK9)是细胞周期蛋白依赖性蛋白激酶家族的成员, 参与多种靶基因的转录延伸, 在促进转录中起着至关重要的作用。已被证明CDK9在胰腺癌、前列腺癌和乳腺癌等多种恶性肿瘤中普遍表达。目前的CDK9抑制剂是可逆的, 并且需要连续占据靶标以维持CDK9抑制, 有一定的局限性。Olson等[15]合成的THAL-SNS-032诱导了CDK9的快速降解, 与另外两种传统的CDK9抑制剂相比, 可以在清除后仍保留细胞毒性作用。不过, 虽然THAL-SNS-032可以特异性降解CDK蛋白, 但仍无法区分降解CDK2、CDK7和CDK9。之后Robb等[16]合成了可以选择性降解CDK9的PROTAC小分子。
1.5 BCR-ABL融合蛋白(breakpoint cluster region-c-abl, BCR-ABL)慢性粒细胞白血病(chronic myelogenous leukemia, CML)通常由致癌融合蛋白BCR-ABL中c-ABL激酶结构域的自动抑制作用的丧失引起, 酪氨酸激酶抑制剂(tyrosine kinase inhibitors, TKIs)是CML有效的治疗药物。有一种理论认为因为BCR-ABL充当补体信号通路中的蛋白质支架, 所以可以使白血病干细胞能够在激酶抑制中存活。因此, 如果可以降解BCR-ABL则可以避免持续用药治疗而治愈CML。Lai等[17]合成了通过劫持CRBN或VHL E3泛素连接酶介导c-ABL和BCR-ABL降解的PROTAC小分子, 靶向降解了可能为支架蛋白的BCR-ABL蛋白。
1.6 B细胞淋巴瘤6蛋白(B-cell lymphoma 6 protein, BCL6)BCL6是体液免疫应答期间生发中心形成和维持所必需的, 但也与源自生发中心的B细胞淋巴瘤的发生发展有关。McCoull等[18]同时合成了BCL6的抑制剂与PROTAC分子, 虽然都具有高活性和良好的细胞杀伤活性, 但对BCL6抑制与降解均未达到显著的选择性作用, 且PROTAC虽然可以有效降解细胞中的BCL6, 但降解并不完全。以BCL6为靶点开发PROTAC分子, 仍存在相应挑战。
迄今为止所构建的PROTAC分子靶点, 多以核内蛋白为主, 以上列出的核内蛋白为近期PROTAC分子研究较多、较有特点的靶点, 除此之外也有以TRIM24[19]、ERRα[20]、Sirtuin 2[21]、HDAC6[22]等为靶点的研究。
2 以跨膜蛋白为靶点的应用现所报道的以跨膜蛋白为靶点的PROTAC分子数量有限, 且大都以受体酪氨酸激酶(receptor tyrosine kinase, RTK)为靶点。与抑制RTK活性的传统策略不同, 激酶的降解也是一种治疗选择, 并且它有可能产生更完整和持久的下游信号失活, 并绕过"kinome重接线"的问题, 即通过替代激酶(gravese)抑制受体信号导致补偿反馈激活。PROTAC分子可以尝试降解RTK而治疗相关疾病, 且有希望比RTK抑制剂的作用更有效持久且对Kinome重接线的敏感性更低。
2.1 间变性淋巴瘤激酶(anaplastic lymphoma kinase, ALK)ALK是一种受体酪氨酸激酶, 属于胰岛素受体激酶亚家族, ALK的活化与多种癌症类型的进展有关, 开发ALK抑制剂以拮抗ALK的激酶活性是治疗策略之一。迄今为止, FDA已批准4种ALK抑制剂用于治疗ALK阳性非小细胞肺癌患者。但是, 这些抑制剂治疗的大多数患者中已有耐药性出现。因此, 需要开发新的治疗方法来延迟或克服耐药性。Zhang等[23]合成两个linker不同的化合物, 均对ALK表现出高亲和力, 且可对ALK融合蛋白可逆降解。Kang等[24]合成了ceritinib与VHL E3泛素连接酶配体形成的TD-004, 不仅有效降低了细胞内融合ALK蛋白的水平, 而且还选择性地抑制了ALK阳性癌细胞SU-DHL-1和H3122的融合增殖。
2.2 上皮生长因子受体(epidermal growth factor receptor, EGFR)EGFR是上皮生长因子细胞增殖和信号传导的受体, 研究表明许多实体肿瘤中存在EGFR的异常表达。EGFR与肿瘤细胞的增殖、血管生成、肿瘤侵袭、转移及细胞凋亡的抑制有关。EGFR的小分子激酶抑制剂可以竞争性地结合到激酶域, 从而阻止信号传导; 抗体能够优先结合同源配体识别位点, 从而阻止激酶活化。此外, FDA批准可降解EGFR的抗体已获得临床成功, 这表明受体的降解可能是有利于治疗疾病的。基于这一基本原理, Crews等[25]以EGFR为靶点, 开发了一种能够诱导EGFR降解的PROTAC小分子, 并证明了PROTAC分子招募元素的不同也可以使蛋白质在不同的突变状态下降解。
2.3 人类表皮生长因子受体2 (human epidermal growth factor receptor-2, HER2)与EGFR相似, HER2的过度表达也是包括卵巢癌、乳腺癌和胃癌在内的多种癌症的致癌因素, Crews等[25]用可降解EGFR的PROTAC分子也对HER2蛋白进行了检测, 实验证明该化合物也可将HER2蛋白降解, 且与激酶抑制相比, 在预防下游信号传导和延迟启动激酶组重新布线方面, PROTAC使RTK降解的效果更好。但此化合物对EGFR与HER2均有降解作用, 并未合成出选择性的HER2降解PROTAC。
2.4 c-间质上皮细胞转换因子(c-mesenchymal-epithelial transition factor, c-Met)c-Met是肝细胞生长因子的受体, 也被称为促进肿瘤转移能力的"分散因子"。c-Met抑制剂foretinib可以竞争性地从c-Met激酶结构域里将ATP取代, 阻断HGF刺激的ERK和AKT的激活。由于肿瘤发生的激酶依赖性的特点, 降解蛋白的治疗效果可能优于抑制。Crews等[25]设计了可降解c-Met的PROTAC分子, 并证明了它对细胞表面成熟的c-Met诱导降解, E3泛素连接酶配体对泛素的募集能够直接或间接诱导蛋白内化。
3 以胞浆蛋白为靶点的应用细胞质是生命活动的主要场所, 进行各种各样的生化反应, 胞浆中存在着多种激酶参与到细胞生命活动调控的各个通路, 并发挥着重要的作用, 它们的异常上调也会导致信号通路的异常, 从而导致疾病发生。
3.1 布鲁顿酪氨酸激酶(Brutons tyrosine kinase, BTK)BTK是B细胞受体信号通路的关键调节因子, 在不同类型恶性血液病中广泛表达, 参与B细胞的增殖、分化与凋亡, 因此被作为治疗恶性血液肿瘤的重要靶点。它的不可逆抑制剂依鲁替尼已成为慢性淋巴细胞白血病和其他B细胞恶性肿瘤患者的转化治疗选择, 但已有较多的患者产生耐药。基于此, Crews等[26]合成了MT-802, 实验表明MT-802相较于依鲁替尼而言, 能够减少活性磷酸化BTK, 可发挥抑制剂所不具有的作用。Huang等[27]合成了可延长药效学作用的选择性BTK降解剂DD-04-015, 并证明了BTK和TEC激酶家族可能特别易受降解策略的影响(表 1)。
| 表 1 The target proteins and structures of small molecule PROTAC |
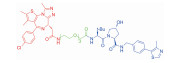
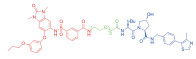
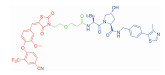
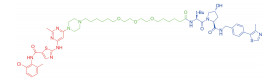
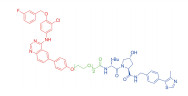
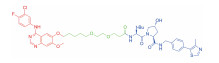
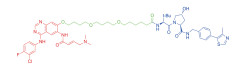
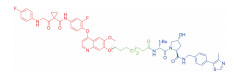
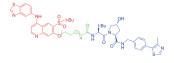


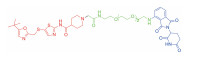
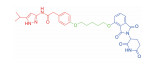
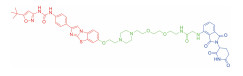
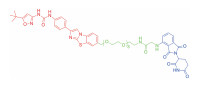
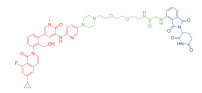
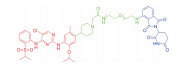
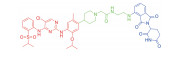
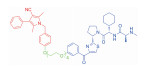

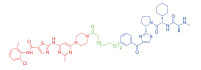
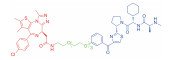
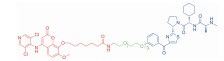
转录因子p53在癌细胞的凋亡中起关键作用, 其失活是肿瘤发生的主要因素。p53的众多功能受MDM2的调节, MDM2的过度表达会导致p53的失活。虽然许多MDM2-p53相互作用的抑制剂已经进展为临床试验, 但是后续研究发现由于药代动力学作用, MDM2抑制剂被清除后, 体内积聚的MDM2蛋白仍可以有效且迅速地降解p53从而降低了MDM2抑制剂的治疗功效, 于是需要新的策略来更有效地靶向降解MDM2[28]。Hines等[29]基于IMiD设计的MDM2 PROTAC 8、王少萌等[28]基于MI-1061设计的MD-224均通过靶向降解有效降低了MDM2蛋白水平, 相对于MDM2-p53抑制剂MI-1061, 也在RS4 11异种移植模型中表现出增强的功效。除作为目标降解的靶蛋白外, MDM2的特殊之处是, 它也可以作为PROTAC的E3连接酶配体部分[6], 且在某些方面上降解能力更强[29]。
4 小结综上所述, 针对在细胞各个分部区域中的靶点都有相应的PROTAC小分子报道, 证明了根据合适的蛋白配体、E3泛素连接酶配体所设计的PROTAC分子可以对核内蛋白、跨膜蛋白、胞浆蛋白有相应的降解作用。然而, 尽管对不同蛋白都有相应的降解作用, 但近年来的研究重点仍只侧重于针对核内蛋白的PROTAC设计, 而对跨膜蛋白、胞内蛋白的研究相对较少。近年来, 针对PROTAC分子的研究越来越多, 但关于靶点选择、分子设计上还是没有完整的体系, 不同定位蛋白研究的深度不一。PROTAC小分子是否有更适合或者不适用的降解靶蛋白的应用, 还需进行进一步的研究。
| [1] |
Sakamoto KM, Kim KB, Kumagai A, et al. PROTACS:chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation[J]. Proc Natl Acad Sci U S A, 2001, 98: 8554-8559. DOI:10.1073/pnas.141230798 |
| [2] |
Salami J, Crews CM. Waste disposal-an attractive strategy for cancer therapy[J]. Science, 2017, 355: 1163-1167. DOI:10.1126/science.aam7340 |
| [3] |
Fei YD, Liu L, Gong Y, et al. PROTAC and its application in the treatment of cancer[J]. Chem Life, 2014, 34: 549-554. |
| [4] |
Duan YC, Zhai XY, Qin WP, et al. Advances in the treatment of cancer by PROTACs[J]. Acta Pharm Sin (药学学报), 2017, 52: 1801-1810. |
| [5] |
Shibata N, Nagai K, Morita Y, et al. Development of protein degradation inducers of androgen receptor by conjugation of androgen receptor ligands and inhibitor of apoptosis protein ligands[J]. J Med Chem, 2018, 61: 543-575. DOI:10.1021/acs.jmedchem.7b00168 |
| [6] |
Schneekloth AR, Pucheault M, Tae HS, et al. Targeted intracellular protein degradation induced by a small molecule:En route to chemical proteomics[J]. Bioorg Med Chem Lett, 2008, 18: 5904-5908. DOI:10.1016/j.bmcl.2008.07.114 |
| [7] |
Han X, Wang C, Qin C, et al. Discovery of ARD-69 as a highly potent proteolysis targeting chimera (PROTAC) degrader of androgen receptor (AR) for the treatment of prostate cancer[J]. J Med Chem, 2019, 62: 941-964. DOI:10.1021/acs.jmedchem.8b01631 |
| [8] |
Chan KH, Zengerle M, Testa A, et al. Impact of target warhead and linkage vector on inducing protein degradation:comparison of bromodomain and extra-terminal (BET) degraders derived from triazolodiazepine (JQ1) and tetrahydroquinoline (I-BET726) BET inhibitor scaffolds[J]. J Med Chem, 2018, 61: 504-513. DOI:10.1021/acs.jmedchem.6b01912 |
| [9] |
Raina K, Lu J, Qian Y, et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer[J]. Proc Natl Acad Sci U S A, 2016, 113: 7124-7129. DOI:10.1073/pnas.1521738113 |
| [10] |
Zengerle M, Chan KH, Ciulli A. Selective small molecule induced degradation of the BET bromodomain protein BRD4[J]. ACS Chem Biol, 2015, 10: 1770-1777. DOI:10.1021/acschembio.5b00216 |
| [11] |
Lu J, Qian Y, Altieri M, et al. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4[J]. Chem Biol, 2015, 22: 755-763. DOI:10.1016/j.chembiol.2015.05.009 |
| [12] |
Winter GE, Buckley DL, Paulk J, et al. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation[J]. Science, 2015(348): 1376-1381. |
| [13] |
Lebraud H, Wright DJ, Johnson CN, et al. Protein degradation by in-cell self-assembly of proteolysis targeting chimeras[J]. ACS Cent Sci, 2016, 2: 927-934. DOI:10.1021/acscentsci.6b00280 |
| [14] |
Ohoka N, Okuhira K, Ito M, et al. In vivo knockdown of pathogenic proteins via specific and nongenetic inhibitor of apoptosis protein (IAP)-dependent protein erasers (SNIPERs)[J]. J Biol Chem, 2017, 292: 4556-4570. DOI:10.1074/jbc.M116.768853 |
| [15] |
Olson CM, Jiang B, Erb MA, et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation[J]. Nat Chem Biol, 2018, 14: 163-170. DOI:10.1038/nchembio.2538 |
| [16] |
Robb CM, Contreras JI, Kour S, et al. Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC)[J]. Chem Commun (Camb), 2017, 53: 7577-7580. DOI:10.1039/C7CC03879H |
| [17] |
Lai AC, Toure M, Hellerschmied D, et al. Modular PROTAC design for the degradation of oncogenic BCR-ABL[J]. Angew Chem Int Ed Engl, 2016, 55: 807-810. DOI:10.1002/anie.201507634 |
| [18] |
McCoull W, Cheung T, Anderson E, et al. Development of a novel B-cell lymphoma 6(BCL6) PROTAC to provide insight into small molecule targeting of BCL6[J]. ACS Chem Biol, 2018, 13: 3131-3141. DOI:10.1021/acschembio.8b00698 |
| [19] |
Gechijian LN, Buckley DL, Lawlor MA, et al. Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands[J]. Nat Chem Biol, 2018, 14: 405-412. DOI:10.1038/s41589-018-0010-y |
| [20] |
Bondeson DP, Mares A, Smith IE, et al. Catalytic in vivo protein knockdown by small-molecule PROTACs[J]. Nat Chem Biol, 2015, 11: 611-617. DOI:10.1038/nchembio.1858 |
| [21] |
Schiedel M, Herp D, Hammelmann S, et al. Chemically induced degradation of sirtuin 2(Sirt2) by a proteolysis targeting chimera (PROTAC) based on sirtuin rearranging ligands (SirReals)[J]. J Med Chem, 2018, 61: 482-491. DOI:10.1021/acs.jmedchem.6b01872 |
| [22] |
Yang K, Song Y, Xie H, et al. Development of the first small molecule histone deacetylase 6(HDAC6) degraders[J]. Bioorg Med Chem Lett, 2018, 28: 2493-2497. DOI:10.1016/j.bmcl.2018.05.057 |
| [23] |
Zhang C, Han XR, Yang X, et al. Proteolysis targeting chimeras (PROTACs) of anaplastic lymphoma kinase (ALK)[J]. Eur J Med Chem, 2018, 151: 304-314. DOI:10.1016/j.ejmech.2018.03.071 |
| [24] |
Kang CH, Lee DH, Lee CO, et al. Induced protein degradation of anaplastic lymphoma kinase (ALK) by proteolysis targeting chimera (PROTAC)[J]. Biochem Biophys Res Commun, 2018, 505: 542-547. DOI:10.1016/j.bbrc.2018.09.169 |
| [25] |
Burslem GM, Smith BE, Lai AC, et al. The advantages of targeted protein degradation over inhibition:an RTK case study[J]. Cell Chem Biol, 2018, 25: 67-77. DOI:10.1016/j.chembiol.2017.09.009 |
| [26] |
Buhimschi AD, Armstrong HA, Toure M, et al. Targeting the C481S ibrutinib-resistance mutation in Bruton's tyrosine kinase using PROTAC-mediated degradation[J]. Biochemistry, 2018, 57: 3564-3575. DOI:10.1021/acs.biochem.8b00391 |
| [27] |
Huang HT, Dobrovolsky D, Paulk J, et al. A chemoproteomic approach to query the degradable kinome using a multi-kinase degrader[J]. Cell Chem Biol, 2018, 25: 88-99. DOI:10.1016/j.chembiol.2017.10.005 |
| [28] |
Li YB, Yang JL, Aguilar A, et al. Discovery of MD-224 as a first-in-class, highly potent, and efficacious proteolysis targeting chimera murine double minute 2 degrader capable of achieving complete and durable tumor regression[J]. J Med Chem, 2019, 62: 448-466. DOI:10.1021/acs.jmedchem.8b00909 |
| [29] |
Hines J, Lartigue S, Dong H, et al. MDM2-recruiting PROTAC offers superior, synergistic antiproliferative activity via simultaneous degradation of BRD4 and stabilization of p53[J]. Cancer Res, 2019, 79: 251-262. DOI:10.1158/0008-5472.CAN-18-2918 |