 2020, Vol. 55
2020, Vol. 55



2. 中国科学院大学, 北京 100049
2. University of Chinese Academy of Sciences, Beijing 100049, China
多肽是由各种氨基酸分子之间脱水形成肽键相连的有机物, 其分子质量在1~10 kDa, 介于小分子和生物大分子之间。存在于体内的诸多信号分子都属于肽或蛋白质, 疾病的发生发展离不开这些肽或蛋白质。就目前已知的活性肽而言, 大部分都是由机体分泌或代谢转化而来。因此, 按照活性肽的来源可以将肽分为两类:第一类是来源于生物体本身的蛋白质及活性肽, 称为内源性活性肽。内源性活性肽在体内含量少、分布广、效应极强。第二类是来源于动植物的活性多肽以及抗生素等, 称为外源性活性肽。外源性活性肽作用强、分布广泛。内源性活性肽和外源性活性肽构成的多肽库为药物研发提供了新颖的结构骨架, 许多上市药物都源自多肽化合物的发现。
但是经典的多肽结构对体内蛋白酶的稳定性较差, 进入体内很快会被降解; 此外, 大多生物活性肽生物利用度比较差, 无法口服, 需要通过改变剂型进而研发获取适合的给药途径。基于以上这些因素, 需要对活性肽进行结构修饰与化学改造[1]。活性肽改造的目的多种多样, 主要包括提高活性肽与受体的亲和力及选择性; 增强多肽分子的药代稳定性, 降低活性肽在体内的降解或者减少活性肽在体内的消除; 提高活性肽的透膜能力; 改善疏水肽的水溶性等。本文针对不同改造目的总结归纳了肽类分子结构修饰改造策略, 根据是否对肽链骨架进行修饰, 将这些修饰策略分为两类:一类是针对肽链骨架的改造, 包括非天然氨基酸修饰、伪肽化策略、逆肽策略、环化策略、末端结构修饰等; 另一类是在多肽骨架不变的基础上, 引入其他基团进行结构优化和性能改造, 包括高级脂肪酸修饰、聚乙二醇修饰、蛋白融合策略、胆固醇修饰等。通过综合运用这些先导化合物结构修饰策略, 能够显著提高多肽类化合物的成药性, 为开发多肽类创新药物提供理论指导和实践经验。
1 提高肽类分子活性药物的化学结构与药理活性之间的关系一直都是药物化学领域的重要研究内容。活性是化合物开发成药物的前提, 多肽也是如此。部分天然肽类分子或人工合成的肽生物活性差, 需要通过化学修饰提高肽类分子与受体的亲和力, 改善肽的活性。提高肽类分子活性的主要方法包括末端结构修饰、拼接策略、环化策略、非天然氨基酸修饰、伪肽策略以及胆固醇修饰等。
1.1 肽链骨架改造对肽链骨架进行修饰和改造以提高肽类分子活性的主要方法包括末端结构修饰、拼接策略、环化策略、非天然氨基酸修饰、伪肽策略等。
1.1.1 N-Cap结构修饰N端裸露和C端裸露的多肽容易受到肽链外切酶的识别, 从而被切割降解失去活性。而将N末端和C末端进行结构修饰, 一方面可以提高肽类分子的代谢稳定性, 另一方面可以保持甚至提高肽类分子的活性。Stoermer等[2]报道了三肽(KKR序列)醛类化合物作为西尼罗病毒(west Nile virus, WNV)蛋白酶抑制剂; 尹正课题组报道了三肽(KRR序列)醛类化合物作为登革热病毒(Dengue virus, DENV)蛋白酶抑制剂, 并且相较于其他四肽醛类化合物活性显著提高[3]。基于此类研究报道, Andreas等[4]认为不同氨基末端结构修饰的三肽醛类化合物对活性有不同影响, 因此他们对N端Cap区进行考察, 得到不同酰基化修饰的三肽醛类化合物, 并且发现不同酰基化修饰对化合物的抗病毒活性有较大影响(表 1)[4], 可以看出N端苯乙酰基修饰的三肽醛2对登革热病毒和西尼罗病毒都有较好的抑制活性, 而N端4-苯基苯乙酰基修饰的三肽醛11相比于2对西尼罗病毒的抑制活性提高近7倍。这一点表明氨基末端的结构修饰对肽类化合物的活性有一定影响, 可以作为肽类化合物改造的一种策略。
| 表 1 N-cap structure modification to affect anti-viral activity against viral protease [4] |
与氨基端结构修饰相对应, 羧基端结构修饰在肽类分子的修饰改造也具有广泛应用, C-末端结构修饰策略成功地在各类病毒蛋白酶抑制剂的结构改造中使用。丙肝病毒NS3/4A蛋白酶是一种丝氨酸蛋白酶, 目前大部分丝氨酸蛋白酶抑制剂都含有亲电基团, 与催化三联体的丝氨酸羟基形成共价键。研究人员从十肽底物出发经过肽链的简化以及羧基末端结构修饰得到活性肽醛19, 其对HCV NS3/4A蛋白酶的结合常数为12 μmol·L-1, 但醛基的化学和代谢稳定性较差。因此, 研究人员对醛基末端进行结构修饰, 以α-卤代酮、杂代酮、α-二酮和α-酮酰胺替换醛基, 得到的酮酰胺化合物20对丝氨酸蛋白酶NS3/4A的结合常数提高了12倍。分析酮酰胺20与丝氨酸蛋白酶的相互作用(图 1), 研究人员发现酮酰胺结构既可以与139位丝氨酸形成共价结合, 又可以与附近的137位谷氨酸和138位丝氨酸残基形成氢键作用, 增强了化合物与NS3/4A蛋白酶的结合, 因而其抗病毒活性提高[5]。

|
图 1 C-terminal structure modification to improve activity against serine protease |
EV71 3C蛋白酶是一种半胱氨酸蛋白酶。尹正等[6]发现了对EV71病毒具有较好抑制活性的肽醛分子21 (EC50=0.11 μmol·L-1), 考虑到醛基的稳定性较差, 成药性质不佳。他们在进一步的结构修饰中, 针对醛基进行结构优化, 得到羟基氰类化合物22, 该化合物对EV71的活性为0.056 μmol·L-1。通过分子对接, 分析化合物22与EV71 3C蛋白酶的相互作用, 分子对接结果表明(图 2), 相比于醛基, 羟基氰结构中的氰基与146位谷氨酰胺和24位谷氨酸通过水分子形成氢键, 增强了化合物与EV71 3C蛋白酶的结合, 因而活性得以提高[6, 7]。

|
图 2 C-terminal structure modification to improve activity against EV71 3C protease. Figure derived from Zhai Y, et al[6] |
在肽类化合物的改造中, 往往需要对不同位点同时进行优化和改造, 拼接策略是一个高效的结构优化方法。首先, 分别对N端和C端结构修饰改造得到活性较优的化合物, 然后将优势片段进行拼接, 即可快速获得活性更高的化合物。拟肽化合物23是一个登革热病毒蛋白酶抑制剂, 其抑制活性IC50为13.3 μmol·L-1。在对该化合物改造的过程中, 研究人员就采用了分别优化N端和C端的研究策略。在N端结构改造中, 研究人员发现肽类分子24, 即N端Cap结构修饰的化合物, 活性提升, 其IC50达到2.5 μmol·L-1。在C端侧链改造过程中研究人员也发现将正丁基侧链替换为苯基得到化合物25, 同样可以提高化合物对登革热病毒的抑制活性, 活性提升近4倍。考虑到这两个修饰策略都可以提高化合物的活性, 研究人员将两个优势片段组合拼接, 得到化合物26, 其对登革热病毒的抑制活性为0.6 μmol·L-1, 活性提高了近20倍(图 3)[8]。

|
图 3 Splicing strategy to improve DENV inhibitory activity |
许多情况下, 直链肽的分子柔性造成构象发生变化, 使其与受体结合的强度及选择性下降。此外, 生物体内的氨肽酶及羧肽酶也易于从直链肽两个端基逐步切割肽链, 使之降解。因此肽链的环化改造, 使其构象限定是改善肽类分子生物稳定性、提高生物活性的重要结构改造策略[9]。研究表明, 从直链肽改为环肽后, 许多化合物的生物活性提高十几倍至几万倍。许多具有抗菌、抗病毒、抗肿瘤、免疫调节等活性的天然产物肽往往含有不同类型的主链环化结构。因此, 环化策略是多肽结构修饰改造的一个重要策略。
线性八肽化合物27对登革热病毒NS2B-NS3蛋白酶有较弱的结合活性(Ki=42 μmol·L-1)。Xu等[10]推测线性肽结合活性较差的原因可能是线性肽占用的空间较大, 无法与蛋白酶有效地结合; 而用环肽则可以改变线性肽所占用的空间, 提高化合物对NS2B-NS3蛋白酶的结合活性。他们设计合成了系列环肽结构, 并且对这类环肽的结合活性进行测试。发现环肽28的构象使得其可以较好地与登革热病毒NS2B-NS3蛋白酶结合, 相较于线性肽活性提高近20倍, Ki值达到2.2 μmol·L-1 (图 4)。

|
图 4 Cyclization strategy to improve activity against NS2B-NS3 protease. Figure derived from Xu SQ, et al[10] |
除了首尾相连的大环化策略, 局部环化往往能够局部限定环化区域的肽类化合物构象, 稳定肽类化合物与受体的相互作用, 提高肽类化合物的活性。信号传导及转录激活因子STAT3是一种直接将细胞外受体的信号传递至核内的转录因子。STAT3的持续激活会促进细胞增殖从而形成肿瘤并且抑制肿瘤细胞凋亡[11-13]。美国密歇根大学王少萌等[14]早期发现gp130 pYLPQTV肽29与STAT3受体有较强的亲和力。研究表明, gp130磷酸肽中的苏氨酸和缬氨酸可以被苄胺替代而不改变肽和STAT3之间的结合活性, 因此, 研究人员切断苏氨酸和缬氨酸并用苄胺封闭碳端, 得到了截断磷酸肽30, 30与STAT3受体的结合力Ki为350 nmol·L-1。通过分子对接, 他们发现亮氨酸侧链异丁基和脯氨酸五元环可以并环形成双环内酰胺结构而不破坏肽30 β转角的构象。因此, 采用局部环化策略, 他们设计合成了构象限制的双环类肽化合物31, 其与STAT3受体的结合力Ki为17 nmol·L-1, 活性提高了近20倍。分子对接结果表明, 双环内酰胺结构可以很好保持30的β转角构象。为了验证Cbz保护基是否和肽31与STAT3受体结合相关, 他们将苄氧羰基替换为乙酰基, 得到的肽32与STAT3受体的结合力Ki值为15 nmol·L-1, 与肽31活性相当, 说明Cbz并非活性必须。随后, 他们评价了肽32对两种含有高磷酸化STAT3受体的人乳腺癌细胞株MDA-MB-231和MDA-MB-468的抑制活性, 但32在100 μmol·L-1水平下对这两种肿瘤细胞并没有表现出抑制活性, 可能是磷酸肽的极性太大, 无法通过细胞膜。为了增强32对肿瘤细胞的抑制活性, 他们将高级脂肪酸引入磷酸肽的氮端, 得到了脂肪酸修饰的磷酸肽33, 33与STAT3受体的结合力Ki值为10 nmol·L-1, 而且肽33对两种细胞的抑制活性IC50分别为25和35 μmol·L-1, 在细胞水平显示了一定的抑制活性, 也表明脂肪酸修饰可以改变肽类化合物的性质, 提高肽类化合物的透膜性(图 5)[15]。

|
图 5 Partial cyclization strategy to improve peptide binding affinity with STAT3 receptor |
卡托普利是第一个报道的血管紧张素转化酶(angiotensin converting enzyme, ACE)抑制剂, 1981年被美国食品药品管理局(FDA)批准用于治疗高血压。临床研究表明, 卡托普利的巯基可能会引起患者皮疹和食欲减退等不良反应。为了解决这一问题, 研究人员研发新型ACE抑制剂作为降压药物。在卡托普利研发早期, 活性化合物34具有一定的ACE抑制活性, 其IC50为4.9 μmol·L-1。对34的亚甲基用氮原子进行生物电子等排, 得到二肽先导化合物35, 其活性提高1倍[16]。由于氮原子的引入, 化合物的亲水性有所增强; 为了平衡氮原子引起的亲水性增强, 研究人员尝试在氨基酸的α位引入烷基侧链平衡亲水性变化, 结果得到的化合物36活性进一步增强至0.09 μmol·L-1。随后, 研究人员对α位烷基侧链进行了详细的构效关系考察, 最终确定苯乙基取代时, 活性最优[17], 将羧基乙酯化开发获得前药依那普利37, 依那普利于1985年被美国FDA批准用于高血压和心力衰竭的治疗。37对ACE的抑制活性相比于36活性提高了74倍, 表明非天然氨基酸的引入可以增强肽类分子的药理活性(图 6)。

|
图 6 Introduction of unnatural amino acids to improve ACE inhibitory activity |
伪肽则是通过模拟多肽水解的过渡态, 利用生物电子等排原理对易水解的酰胺键进行替换, 使多肽免于蛋白酶的水解切割从而保留甚至提高肽类化合物的药理活性。图 7列出了一些伪肽的代表结构[18]。

|
图 7 Representative structures of pseudopeptides |
片段38 (羟基亚甲基)是众多HIV蛋白酶抑制剂、肾素抑制剂和β-分泌酶抑制剂[19]共有的结构片段。其基本的设计原理就是利用伪肽策略, 模拟酰胺键水解过程中的过渡态, 替换易水解的酰胺键。Szelke等[20]通过在肾素底物42的亮氨酸-缬氨酸(Leu-Val)片段中采用羟基亚甲基替换酰胺键, 得到伪肽抑制剂43, 对HIV-1蛋白酶抑制活性显著提高(图 8), 其IC50值为0.000 7 μmol·L-1。

|
图 8 Pseudopeptide strategy to improve anti-HIV-1 protease activity |
其中缩硅酮片段41也有广泛应用, 由于碳原子和硅原子同属一个主族, 两个原子的性质十分相似, 而硅原子相比碳原子更倾向于sp3杂化, 片段41不容易发生消除反应生成硅酮, 缩硅酮的稳定性要高于缩酮, 因此在设计和改造活性肽的时候引入41片段既可以增强与酶活性中心的相互作用, 又具有一定的化学稳定性, 在药物化学化合物设计中具有广泛应用(图 9)。片段41在很多活性肽类似物分子上显示出良好活性, 例如含有片段41的ACE抑制剂44, 其对ACE酶的抑制活性达到了3.8 nmol·L-1; 而含有片段41的HIV蛋白酶抑制剂45对蛋白酶的抑制活性也达到2.7 nmol·L-1, 表明该类结构在活性肽结构改造中有重要意义[21]。

|
图 9 Representative silicon containing pseudopeptide inhibitors |
外接基团修饰以提高肽类分子活性的主要方法是胆固醇修饰。
胆固醇修饰也是多肽类分子的重要结构修饰策略。胆固醇的引入常常可以在提高其在体内半衰期的同时增强多肽的药理活性。Wang等[22]用细胞-细胞融合实验评价多肽分子的抗病毒活性, 发现多肽m4HR具有一定抗HIV-1活性(IC50 =36 910 nmol·L-1)。当在m4HR C末端外接胆固醇分子得到化合物46, 其抗病毒活性提高200倍(IC50 =57.2 nmol·L-1)。进一步在N端修饰, 得到的肽类化合物对HIV-1的抑制活性进一步提升(表 2)。其中活性最好的是肽类分子47, 其IC50达到8.3 nmol·L-1, 该类化合物结构优化的实例进一步证明了胆固醇修饰在多肽药物活性优化的重要应用。
| 表 2 Cholesterol conjugation to improve anti-viral activity against HIV-1 |
多肽的基本组成单元是氨基酸, 其本质与蛋白质相同, 因而多肽类分子是许多蛋白酶水解的底物, 而这一特点严重限制了多肽类药物的开发研究。一般而言, 大部分多肽类药物无法口服, 否则就会被胃蛋白酶以及胰蛋白酶等消化破坏; 其次, 即使通过注射给药, 多肽类药物也有可能在血液以及组织中被蛋白酶降解失活, 因此多肽类药物的生物利用度很低, 以至于多肽类分子在临床治疗中受到很大限制[23]。为了减弱或避免蛋白酶对多肽类分子的降解, 必须要利用化学方法或其他方法对多肽分子进行修饰改造, 以提高多肽的代谢稳定性, 为新药研发中解决多肽的代谢稳定性问题提供一些思路和参考。增强多肽分子代谢稳定性的主要方法包括非天然氨基酸修饰、伪肽化策略、逆肽策略、环化策略以及高级脂肪酸修饰、蛋白融合策略、聚乙二醇修饰等。
2.1 肽链骨架改造对肽链骨架进行修饰和改造以增强多肽分子代谢稳定性的主要方法包括非天然氨基酸修饰、伪肽化策略、逆肽策略、环化策略等。
2.1.1 非天然氨基酸修饰天然活性肽的组成常常都是天然氨基酸。天然活性肽容易受到体内蛋白酶降解, 从而降低其在体内的半衰期, 导致天然活性肽在体内发挥药效时间缩短, 不利于成药。β氨基酸作为非天然氨基酸, 在体内不易被蛋白酶识别水解, 在活性肽的结构改造与修饰中发挥重要作用。
化合物51是一个神经降压素, 其作用于神经降压素受体1和2 (NTSR1和NTSR2)两个亚型。神经降压素及其受体与痛觉缺失的调节、食物摄取以及肿瘤生长具有密切关系[24]。研究人员对神经降压素51进行结构优化, 经过截断策略得到了含有第八位到第十三位氨基酸序列的简化肽52 (NTSR1 Ki=0.24 nmol·L-1; NTSR2 Ki=1.2 nmol·L-1), 其对NTSR1和NTSR2受体的活性均比神经降压素51有所提高。然而, 简化肽52容易受到体内酶代谢作用, 因此其在体内半衰期很短。针对这一特点, 研究人员尝试引入β氨基酸(图 10), 得到活性肽53, 其对NTSR1和NTSR2受体的活性虽然下降(NTSR1 Ki =8 nmol·L-1; NTSR2 Ki=25 nmol·L-1), 但半衰期延长至32 h。之后研究人员又将N端的精氨酸替换为β-精氨酸得到肽54。54相比于53活性略有提高(NTSR1 Ki =6 nmol·L-1; NTSR2 Ki=12 nmol·L-1), 而且54的半衰期大于7天[25, 26], 极大地提高了活性肽在体内的停留时间, 增强了活性肽在体内的药代稳定性。

|
图 10 Introduction of β-amino acids to improve the metabolic stability of peptides |
肽类小分子55是一个广泛研究的金属蛋白酶EP24.15 (endopeptidase)抑制剂。EP24.15与下丘脑对垂体功能的调节及血压调节有重要关联, 文献报道EP24.15还可能与Aβ蛋白的聚集和阿尔兹海默症(Alzheimer's disease, AD)相关, 因此EP24.15是精神系统疾病的研究热点。虽然肽类小分子55对EP24.15的抑制活性很强(IC50=0.06 μmol·L-1), 但它容易受到与EP24.15相关的蛋白酶——中性内肽酶EP24.11水解。因此, 研究人员的主要研发目标是提高55对中性内肽酶EP24.11的稳定性。他们尝试将55中的丙氨酸、酪氨酸和羧基末端分别用β-丙氨酸、β-苯丙氨酸和β-氨基丙酸替换, 得到β肽56对EP24.15的抑制活性虽然有所下降(IC50=2.8 μmol·L-1), 但对中性内肽酶EP24.11的稳定性显著提高(图 11), 几乎不受其降解影响[27]。

|
图 11 Modification of 55 with β-amino acids to yield an inhibitor with complete stability against EP24.11 |
研究人员用图 12的示意图解释引入β氨基酸可以提高肽类分子对中性内肽酶的稳定性。对于天然α多肽, 在特异性的蛋白酶切割位点, 水分子首先与酰胺键形成氢键作用, 从而有利于水分子对酰胺键的进攻最后完成酰胺键的切割; 对β多肽而言, 由于增加了一个亚甲基, 多肽整体的构象发生变化, 原本蛋白酶切割中心的水分子无法与酰胺键形成氢键, 不利于蛋白酶对酰胺键的切割, 因而β多肽比α多肽具有更强的抗水解能力[28]。

|
图 12 Schematic diagram of how a β-peptide may not be cleaved by the peptidase |
阿片受体与疼痛密切相关, 主要包括μ受体、δ受体和κ受体等几种亚型。阿片肽则是一种内源性神经递质, 通过与这些受体结合发挥药理作用。研究人员发现内吗啡肽57是μ受体的内源性底物肽, 具有较强的药理活性, 其对μ受体的激动活性为14.40 nmol·L-1, 相较于吗啡不会产生严重的不良反应; 而且, 内吗啡肽在有效剂量下不易诱发呼吸抑制和心血管疾病。因此, 内吗啡肽引起了科学家的广泛关注。然而, 内吗啡肽仍存在一些问题, 其中之一就是其代谢稳定性较差, 半衰期仅为16.9 min。兰州大学王锐团队发现含有非天然氨基酸的内吗啡肽类似物具有较强的代谢稳定性(图 13), 而且可以在一定程度上进一步提高内吗啡肽对μ受体的活性。他们首先将C末端苯丙氨酸替换为非天然氨基酸, 得到化合物58, 其对μ受体的激动活性为0.033 4 nmol·L-1, 相比于内吗啡肽57提高了430倍; 而且该化合物在脑膜匀浆中的半衰期延长至85.9 min, 与内吗啡肽相比提高了近4倍[29]; 随后, 他们在此工作的基础上进一步把酪氨酸和脯氨酸片段用非天然氨基酸替换, 得到化合物59, 其对μ受体的活性进一步提高, 达到0.042 0 pmol·L-1。而且化合物59在脑膜匀浆中的半衰期超过600 min[30], 解决了内源性吗啡肽半衰期短的问题。因此, 非天然氨基酸的引入对改善肽类化合物的代谢稳定性具有重要意义。

|
图 13 Introduction of unnatural amino acids to improve the stability of endomorphin and its potent analogues |
天然多肽大多由L型氨基酸组成, 容易受到各种蛋白酶的降解而失去活性。蛋白酶的水解反应一般都是立体专一的, 引入D型氨基酸使多肽的构型发生变化, 进而使得修饰的多肽不易被蛋白水解酶水解, 因此D型氨基酸修饰的多肽可以提高对蛋白酶的降解作用。
黄体激素释放激素(luteinizing hormone releasing hormone, LHRH)是由下丘脑分泌的具有调节生殖功能的十肽, 该激素与垂体前叶的黄体激素释放激素受体(gonadotropin-releasing hormone receptor, GnRHR)结合, 可以调控黄体激素的合成和分泌。除此之外, 在人类多种恶性肿瘤中, LHRH与其他生长因子一起调节肿瘤细胞生长。LHRH及类似物可以通过抑制垂体-性腺轴的功能从而抑制激素依赖性肿瘤细胞的增殖, 因此LHRH及类似物目前在临床上用于治疗激素依赖性肿瘤如前列腺癌和乳腺癌等。然而天然的LHRH第5、6位以及第6、7位氨基酸残基间肽键稳定性较差, 在体内极易受到肽链内切酶的作用而裂解, LHRH在体内的半衰期仅有2~4 min。为了提高LHRH在体内的稳定性, 研究人员尝试在6位引入不同种类的D型氨基酸, 得到上市药物如那法瑞林60和曲普瑞林61, 半衰期相较于LHRH均有不同程度的提高, 其半衰期分别为3 h和4 h (表 3)[31]。
| 表 3 Introduction of D amino acids to improve the stability of peptides |
肽键(-CONH2-)是肽类分子的特征, 而肽键在体内容易被蛋白酶识别降解, 这是肽类分子稳定性差的原因之一。伪肽则是利用生物电子等排原理将肽键中的一种或两种以上的原子用其他原子替代。由于伪肽从本质上改变了酰胺键的化学结构, 与蛋白或多肽同源结构不同, 因此可以避免体内蛋白酶的识别和水解, 从而提高肽类分子的稳定性及活性。
N-甲基-D-天冬氨酸(N-methyl-D-aspartate, NMDA)受体与其胞内突触后致密蛋白(postsynaptic density protein-95, PSD-95)的蛋白-蛋白相互作用是治疗缺血性脑病、神经疼痛以及阿尔兹海默症的一种潜在策略[32]。Bach等[33]报道, N-烷基化的谷氨酸-苏氨酸-丙氨酸-缬氨酸四肽化合物(N-甲基-ETAV) 62是NMDA/PSD-95蛋白-蛋白相互作用抑制剂(Ki=9.65 μmol·L-1), 他们通过结构修饰得到一系列活性较强的四肽衍生物, 但是研究人员在改造过程中发现这类化合物的血浆稳定性较差, 例如, 化合物62在人血浆中的半衰期只有113 min, 较差的代谢稳定性限制了该类化合物的进一步开发。为了改善化合物的血浆稳定性, Bach等对该类化合物进行伪肽化结构修饰, 将酰胺键的氧原子用硫原子进行替换, 得到不易被蛋白酶识别并水解的硫杂酰胺键。比较含硫杂酰胺键的伪肽63、64和含有酰胺键的化合物62, 可以发现含硫杂酰胺键的化合物虽然活性有所下降, 但血浆稳定性显著提高(图 14), 尤其是化合物63, 在活性基本不变(Ki=10.8 μmol·L-1)的同时血浆半衰期提高了50倍。研究结果表明, 硫杂酰胺键的伪肽化修饰是提高肽类化合物血浆稳定性的有效策略。

|
图 14 Thionamide pseudopeptides to improve stability of peptides |
蛋白质、激素、活性肽以及天然产物多肽是各种蛋白酶降解的底物, 因此存在着易受蛋白酶降解以及半衰期较短的特点。除了之前介绍的策略可以有效耐受蛋白酶的水解, 肽键方向的改变同样可以改变蛋白酶对底物的识别作用, 从而达到抗降解的作用。这类改变肽键方向的多肽结构修饰策略称为逆肽化修饰, 相关的肽称为逆肽或逆反肽。
β-淀粉样蛋白(amyloid β-protein, Aβ)沉积物的形成可能是引起AD的重要过程。研究表明Aβ可溶性寡聚体有细胞毒性, 并且对大脑的记忆能力和学习能力具有潜在影响。在Aβ聚集的早期进行抑制可有效治疗AD。Taylor等[34]报道了能有效抑制Aβ聚集的九肽65, 尽管65对Aβ寡聚体的聚集有较强的抑制作用, 但65存在多个水解位点, 因此需要对65进行结构修饰以提高其代谢稳定性。对65进行逆肽修饰, 得到逆肽66, 理论上逆肽可以保持与65相似的三维结构从而使活性得到保持, 实验结果也表明逆肽66对Aβ寡聚体的聚集抑制活性并没有发生显著变化。Taylor等用蛋白质降解实验评价65和66的代谢稳定性, 即将肽与人血浆或脑提取物共孵育24 h, 通过高效液相色谱法(HPLC)测定溶液中原型肽含量。可以发现无论血浆还是脑提取物中, 逆肽66的含量均远远高于65, 而且65的含量接近100%, 表明逆肽可以一定程度上提高化合物的代谢稳定性(图 15)。

|
图 15 Inverse-peptide strategy to improve stability of peptides. Figure derived from Taylor M, et al[34] |
肽去甲酰基酶(peptide deformylase, PDF)是参与细菌蛋白质生物合成和成熟的重要酶, 在细菌和真核生物的细胞器中, 蛋白质的合成始于N-甲酰蛋氨酸, 因此新合成的多肽都含有甲酰化的N末端。PDF催化这些多肽的去甲酰化过程。PDF在细菌细胞中发挥的重要作用使其成为设计新型抗生素, 治疗耐药性病原体的新靶标。研究人员在前期工作基础上发现化合物67具有一定的抗菌活性, 其对大肠杆菌PDF抑制活性Ki值为92 nmol·L-1。但67在大鼠血浆中容易受到类胰蛋白酶的降解作用而失活。从图 16中可以发现, 67在大鼠血浆中孵育5 h约25%被降解。为了提高血浆稳定性, 研究人员将P1'与P3'进行环化, 设计合成环肽类似物68。研究结果表明, 相比于67, 环肽类似物68的抗菌活性有所提高, Ki值为74 nmol·L-1, 而且血浆稳定性大幅提高, 将68与大鼠血浆孵育5 h基本不被降解[35]。

|
图 16 Cyclization strategy to improve stability of peptides. Figure derived from Hu XB, et al[35] |
α螺旋是大部分多肽分子都具有的二级结构特征, 然而人工合成的多肽分子在水溶液中并不能保持稳定的α螺旋结构[36], 因此科研人员开发了一种以碳-碳键或其他连接链为支撑的骨架稳定多肽α螺旋结构, 由这类方法得到的多肽称为订书肽(stapled peptide), 该方法本质上也属于环化修饰策略的一种。线性肽柔性大, 在舒展的构象下, 容易暴露出更多酶解位点, 增加了多肽被水解的概率, 从而导致多肽稳定性降低[37]。形成订书肽可以约束线性多肽的构象, 减少多肽被降解的概率。
β连环蛋白-B细胞淋巴瘤9 (B-cell lymphoma, BCL9)蛋白-蛋白相互作用对β连环蛋白的转录活性至关重要, 而这一相互作用是由BCL9蛋白中25个残基的螺旋片段和β连环蛋白的结合槽介导。王少萌等发现, 372位突变的BCL9肽69具有一定抑制β连环蛋白的活性(Ki=0.94 μmol·L-1), 然而69稳定性较差, 在细胞培养液中1 h降解75% (图 17)。因此王少萌等设计了一类结构稳定, 不容易被代谢的BCL9肽[38]。在设计过程中, 他们采用了点击化学(click chemistry)形成的三氮唑为支撑结构, 合成订书肽70和71。订书肽70和71对β连环蛋白的抑制活性分别为0.61 μmol·L-1和0.19 μmol·L-1, 活性保持。同时提高了线性肽的稳定性, 70和71在细胞培养液中1 h仅分别降解30%和25%。

|
图 17 Click chemistry mediated stapled peptide to improve stability of peptides. Figure derived from Kawamoto KA, et al[38] |
外接基团修饰以增强多肽分子代谢稳定性的主要方法包括高级脂肪酸修饰、蛋白融合策略、聚乙二醇修饰等。
2.2.1 高级脂肪酸修饰高级脂肪酸修饰是指在肽类药物的特定位点通过化学方法以共价键的形式引入高级脂肪酸以改善肽类药物的性质, 延长半衰期。一般认为, 高级脂肪酸修饰可以稳定其结构, 提高多肽的稳定性, 从而延长多肽药物在体内的半衰期。同时, 高级脂肪酸与细胞膜表面的磷脂结构类似。因此, 脂肪酸修饰的多肽药物往往也可以提高多肽药物的脂溶性, 改善药物在肠道内的吸收以及黏膜透过性。此外, 高级脂肪酸可以与血清白蛋白(human serum albumin, HSA)结合, 结合后的复合体因分子过大而不容易转运, 从而可以延长多肽在体内的循环时间[39]。目前, 高级脂肪酸作为修饰结构的研究发展仍然比较缓慢, 但高级脂肪酸作为体内的一种内源性物质, 一直吸引了研究人员的广泛关注。根据水蛭素结构简化得到的水蛭肽比伐卢定72 (Bivalirudin)是由The Medicines Company开发的抗凝药物, 于2000年12月年被FDA批准上市, 作为抗凝剂用于经皮冠状动脉腔内成形术(percutaneous transluminal coronary angioplasty, PTCA)治疗中出现的不稳定型心绞痛和经皮冠状动脉介入治疗(percutaneous coronary intervention, PCI)。但是作为多肽类药物, 72在体内的暴露量较低(AUC0-t为23.7 nmol·min·mL-1), 半衰期短(t1/2 =15.1 min), 药代动力学性质较差。在进行经皮冠状动脉介入治疗之前, 需要先进行静脉注射, 随后静脉滴注至手术结束, 患者依从性差。针对这一缺点, 研究人员对比伐卢定类似物73进行化学修饰, 主要的策略是用高级脂肪酸对氨基酸侧链进行修饰。对比肽73和74, 其药理活性基本保持不变, 而高级脂肪酸修饰的多肽74暴露量(AUC0-t为1371.7 nmol·min·mL-1)和半衰期(t1/2=212.2 min)相较于未修饰的多肽73 (AUC0-t为25.7 nmol·min·mL-1, t1/2=13.5 min)明显改善(表 4), 暴露量和半衰期分别提高了58倍和14倍[40]。
| 表 4 Introduction of fatty acid to improve the pharmacokinetic properties of bivalirudin analogs |
上市的降糖多肽药物利拉鲁肽[41]和索马鲁肽[42]也都引入了高级脂肪酸修饰, 高级脂肪酸的引入增加了药物的疏水性, 掩盖二肽基肽酶4 (DPP-4)的结合位点, 降低肾排泄, 提高半衰期。利拉鲁肽是由诺和诺德公司研发的长效GLP-1受体激动剂, 其与天然GLP-1有97%的氨基酸序列相似性, 仅在34位将赖氨酸替换为精氨酸, 同时在26位赖氨酸侧链引入由谷氨酸作为linker的16碳棕榈酸侧链。皮下注射利拉鲁肽后, 其可在注射部位形成稳定的七聚体, 在皮下组织缓慢吸收; 另外, 由于引入了长链脂肪酸修饰, 掩盖了DPP-4结合位点; 同时, 长链脂肪酸的引入还使利拉鲁肽与血清白蛋白形成可逆复合物, 极大地延长了利拉鲁肽在体内的吸收时间, 提高了多肽类药物的体内半衰期。天然的GLP-1半衰期极短, 只有2 min左右; 而棕榈酸修饰的利拉鲁肽半衰期延长至13 h, 提高了390倍(图 18)[43, 44]。索马鲁肽则是GLP-1(7-37)的第8位丙氨酸用氨基异丁酸替换, 34位的赖氨酸用精氨酸替换, 同时在26位赖氨酸侧链由谷氨酸作为linker引入十八烷酸, 疏水性也更强, 同时经过短链的聚乙二醇修饰, 其半衰期大大延长至一周。

|
图 18 Introduction of fatty acid to improve the half-life of GLP-1 analogues |
除了利用可逆的结合方式结合HSA, 共价不可逆的结合方式也常常用于多肽类药物的改造中。艾博卫泰(albuvirtide)是由南京前沿生物技术有限公司开发的全球首个长效抗HIV-1药物, 其结构如图 19所示。恩夫韦肽(enfuvirtide)是FDA批准的第一个临床使用的HIV-1融合抑制剂。然而恩夫韦肽75作为一个多肽药物, 其在人体内的半衰期只有3.5~4.4 h, 需要每天注射两次, 患者的依从性较差。针对恩夫韦肽75半衰期较短的缺点, 前沿生物技术有限公司开发了艾博卫泰, 艾博卫泰76是在多肽序列13位的赖氨酸侧链中引入了3-马来酰亚胺-丙酸(MPA)修饰(图 19), MPA可与血清白蛋白中的巯基形成不可逆的共价结合, 而且结合速率快, 大大提高了多肽在人体内的半衰期, 给药频率一周一次即可[45]。艾博卫泰已于2018年获得国家食品药品监督管理总局(CFDA)批准上市, 用于与其他抗逆转录病毒药物联合使用, 治疗HIV-1感染。

|
图 19 Introduction of MPA to improve the half-life of anti-HIV-1 drug |
蛋白融合策略是指利用基因工程技术, 将蛋白或多肽分子与免疫球蛋白Fc片段或血清白蛋白HSA融合而产生新型分子的修饰策略。融合Fc或HSA片段之后的多肽分子, 分子尺寸显著增大, 降低了肾对多肽药物的清除率, 从而延长多肽药物的半衰期[46]。礼来公司开发的降糖药物度拉糖肽(dulaglutide)就是将GLP-1与IgG4 (Fc)融合而成的长效降糖药物[47], 其生物半衰期大于90 h, 并且疗效不弱于利拉鲁肽, 其在2019年前三季度的销售额达到29.20亿美元, 超过利拉鲁肽(2019年前三季度销售额为24.47亿美元)。
人血清白蛋白是血浆中含量最丰富的蛋白质, 其半衰期长达19天, 因此HSA蛋白融合可以延长多肽药物的半衰期。葛兰素史克公司研发的长效降糖药物阿必鲁肽(albighztide)是第一个被FDA批准上市的HSA蛋白融合药物, 阿必鲁肽的半衰期长达6~10天[48]。因此, 蛋白融合策略是多肽药物长效化的有效手段。
2.2.3 聚乙二醇修饰聚乙二醇(polyethylene glycol, PEG)在体内具有可降解、低毒性、无抗原性等特点, 是一种常见的肽类分子修饰方法。PEG修饰可以改善肽类分子的稳定性、减少蛋白酶的降解、不易被肾小球滤过, 从而提高多肽药物的稳定性, 延长药物的半衰期。目前已有诸多PEG修饰的多肽药物上市, 其中PEG修饰的干扰素α是这一结构修饰策略的成功案例。干扰素α可以有效地抑制或清除乙型肝炎或丙型肝炎病毒, 但干扰素α作为多肽类药物具有自身不可克服的缺点, 其半衰期短, 仅为4 h, 需要每天注射一次。为了克服这一缺点, 先灵葆雅研究所(Schering-Plough Research Institute, SPRI)致力于长效干扰素α的研究。他们分析了干扰素α半衰期较短的原因, 认为其分子过小容易被肾脏清除, 因此研究人员将PEG引入到干扰素α中(图 20), 而修饰后的干扰素α整体分子尺寸变大, 不易被肾小球滤过, 从而PEG修饰的干扰素α半衰期得到延长, 达到40 h[49]。另一方面, 由于PEG的引入掩盖了干扰素α与受体的结合, 降低了干扰素α的抗病毒活性, 因而先灵葆雅研究所对PEG的尺寸进行考察, 最终确定PEG的大小为12 kDa可以在延长半衰期的同时最大程度保留了干扰素α的抗病毒活性。因此, 采用PEG修饰策略要注意平衡半衰期和活性的关系。

|
图 20 PEG modification to improve half-life of interferon-α |
除了少数疏水肽, 大部分多肽都具有极性侧链; 同时, 多肽分子中的肽键可以与水分子形成氢键。因此, 大部分多肽都具有很好的水溶性。而多肽药物必须透过细胞膜才能吸收入血, 发挥药理学活性。因此, 必须要对多肽进行结构修饰与改造, 提高肽类分子的渗透性, 以利于多肽分子进入细胞, 发挥活性。提高肽类分子渗透性的方法包括引入卤素原子、去除极性侧链、手性策略、N-烷基化、高级脂肪酸修饰和其他方法等。
3.1 引入卤素原子在小分子药物的化学修饰中, 往往采用引入卤素的修饰策略以提高小分子药物的亲脂性。在肽类分子的修饰改造中, 卤素的引入也可以提高肽类分子的脂溶性。神经多肽内吗啡肽具有很强的镇痛活性, 然而内吗啡肽57作为一种肽类分子, 很难通过血脑屏障进入大脑发挥药效。通常药物分子进入血脑屏障需要一定的亲脂性, 兰州大学王锐等采用了引入卤素原子的策略, 将2位脯氨酸变换为D-丙氨酸, 4位苯丙氨酸上引入卤素以提高整体肽分子的脂溶性[50], 通过该策略可以明显提高内吗啡肽类似物的渗透性, 可以通过血脑屏障。内吗啡肽57的脂水分配系数D仅有12.5, 而引入卤原子, 内吗啡肽类似物的D值上升至120, 提升了近9倍(图 21)。通过动物实验, 脑实质中检测到了肽77, 进一步验证了通过引入卤素原子可以使肽类分子通过血脑屏障[51]。

|
图 21 Introduction of halogen to improve permeability of endomorphin |
肽类分子中常含有极性的羧基片段, 这些富含谷氨酸和天冬氨酸的肽细胞渗透性比较差, 针对这类肽分子的改造一般采用去除极性侧链的策略。一方面, 去除极性侧链可以缩小肽分子的尺寸, 降低肽链多肽的性质, 使其更具有类似有机小分子的性质; 同时也可以改善肽类分子的细胞渗透性, 有利于其进入细胞发挥药效。比较典型的案例就是抗丙肝病毒药物特拉匹韦的研发(图 22)。Vertex公司早期发现了底物十肽78的活性为0.89 μmol·L-1, 然而该化合物的分子量大, 需要首先对分子大小进行优化。研究人员考察了去除不同氨基酸片段对化合物抗病毒活性的影响, 研究表明去除P4'氨基酸片段对活性影响较大, 而去除P2'和P3'氨基酸片段对酶的亲和力几乎无影响。去除P5和P6两个含有酸性侧链的氨基酸片段, 活性明显降低。另外, 去除P3和P4两个疏水性氨基酸片段也会导致活性的下降。同时考虑到对丝氨酸蛋白酶的结合能力, 研究人员在C末端引入亲电性的醛基作为弹头, 得到了跨越S6~S1的六肽醛79, 其活性与底物十肽相同, 但是分子量显著降低。

|
图 22 Optimization of anti-HCV drug telaprevir |
虽然P5和P6两个酸性氨基酸片段对活性很重要, 但是由于两个羧基的存在, 六肽醛79的极性很大, 不利于化合物进入病毒感染的细胞, 因此下一步结构改造的重点是提高分子的透膜性。研究人员进一步切断P5和P6两个氨基酸片段, 并以杂环进行替换得到四肽醛19, 其抗病毒活性明显下降, 但是相比于底物十肽78, 分子量减小一半, 整个分子的成药性更好[52]。由于醛基的代谢性质较差, 因此醛弹头被其他弹头替换得到酮酰胺化合物20, 其抗病毒活性与稳定性俱佳, 至此, 肽类分子的透膜性问题得以解决。
化合物20的活性仍有提高的空间, 因此研究人员对P1~P4片段进行了系统性优化。他们发现脯氨酸的疏水性基团对酶的亲和力影响很大, 因此首先对P2片段进行改造, 通过比较不同的疏水基团如醚、酯以及氨基甲酸酯等衍生物, 最后得到的含有四氢异喹啉氨基甲酸酯衍生物80, 其对NS3/4A酶的抑制活性提高至0.22 μmol·L-1。进一步优化P1, 发现S1口袋仅能容纳尺寸较小的疏水性基团, 最终确定乙基侧链为最优; 同时优化P3和P4, 得到的化合物81活性与80相当, 但是81的cLogP (5.5)更符合Linpinski五规则, 因而对81进一步研究。将P2乙基侧链与脯氨酸环化, 进一步考察最终确定化合物82, 其对丙肝病毒NS3/4A蛋白酶的抑制活性为44 nmol·L-1, 是一个活性很高的丝氨酸蛋白酶抑制剂, 被命名为特拉匹韦[53]。特拉匹韦于2011年被FDA批准上市, 用于治疗丙型肝炎病毒感染。
3.3 手性策略环肽类化合物的二级结构与其理化性质和药理学性质密切相关。北京大学深圳研究院李子刚等提出了一种假设——在约束肽的连接链上引入一个手性中心以改变肽类分子的理化性质和二级结构, 从而影响肽类分子的透膜性。为验证这一策略的合理性, 他们设计合成了两条FITC标记的含有手性中心的环肽化合物83和84。由于手性中心的存在, 环肽83和84存在一对非对映异构体, 分离出这些异构体83a/b和84a/b并且将之与HEK293T细胞于37 oC共孵育2 h, 用荧光共聚焦显微镜成像(图 23)。研究结果表明, 其中一种构型的异构体83b和84b可以穿入HEK293T细胞, 而另一构型的异构体83a和84a无法穿入细胞。说明手性中心的引入可以使肽的螺旋结构发生变化, 从而影响肽类分子的透膜性[54]。

|
图 23 Introduction of chiral center in cyclopeptide linker to change permeability of peptide. Figure derived from Hu K, et al[54] |
N-烷基化的酰胺键往往可以改变肽类分子内或分子间的氢键相互作用, 从而影响肽类分子的空间结构进而改变其物理化学性质。柔性肽类分子中的分子内氢键是被动扩散中的决定性因素。通过在特定的酰胺键进行烷基化, 可以使肽类分子以最优势的构象穿过细胞膜[55]。Beck等[56]对丙氨酸环六肽进行N-甲基化修饰以考察N-甲基化对丙氨酸环六肽透膜性的影响。实验结果表明, 在1, 5位、1, 6位或1, 2, 4, 5位酰胺氮原子进行甲基化修饰可以明显提高肽类分子对Caco-2细胞的渗透性, 其渗透性与对照睾酮(细胞透膜性标志物)相当。分析1, 6位N-甲基化修饰的环六肽的空间构象发现, 2位酰胺氢与5位酰胺羰基可以形成分子内氢键, 而3位4位氨基酸所形成的β转角也形成了分子内氢键, 整个分子以疏水的构象存在(图 24), 因而细胞渗透性提高。

|
图 24 N-alkylation to improve permeability of peptides. Figure derived from Beck JG, et al[56] |
提高肽类分子透膜性的常用策略是对多肽进行高级脂肪酸修饰。脂肪酸包括不饱和脂肪酸和饱和脂肪酸, 目前有一些饱和脂肪酸修饰的多肽药物已经上市用于疾病的治疗, 或者开发处于临床研究阶段。脂肪酸是构成磷脂双分子层以及人体脂肪的重要成分, 因此对多肽进行脂肪酸修饰可以提高多肽与细胞膜表面的亲和能力, 从而提高肽类分子的透膜性, 促进上皮细胞对肽类分子的吸收。Hashizume等[57]对胰岛素分子的侧链进行棕榈酸修饰, 棕榈酸酰化胰岛素的亲脂性提高。研究人员用同位素标记胰岛素, 并通过测定给药后6 h内血浆中的放射性推断胰岛素在血浆中的含量。结果表明, 双棕榈酰胰岛素的含量最高时是天然胰岛素的6倍, 单棕榈酰胰岛素的含量是天然胰岛素的3倍(图 25)。这也说明高级脂肪酸修饰可以提高肽类分子的透膜性。

|
图 25 Introduction of fatty acid to improve the permeability of insulin. Figure derived from Hashizume M, et al[57] |
除了化学方法, 某些制剂手段也可以影响肽类化合物的渗透和吸收。N-[8-(2-羟苯基)氨基]辛酸钠(sodium N-[8-(2-hydroxybenzyl)amino]caprylate, SNAC)是由Emisphere开发的一种基于各种促吸收剂的大分子递送技术。SNAC能够递送0.5~150 kDa的大分子, 且不会影响大分子的高级结构, 不影响药物释放。同时SNAC还具有很高的安全性, 不影响胃肠黏膜结构。
吸收促进剂与药物分子存在较弱的非共价相互作用, 可形成暂时稳定的中间体。促进剂一般是疏水性物质, 通过与药物分子相互作用形成的药物促进剂复合体具有更强的亲脂性, 从而促进药物分子透过上皮细胞膜。由于复合体只存在较弱的非共价相互作用, 随着复合物透过细胞进入血液循环, 药物与促进剂解离释放出药物(图 26)[58]。

|
图 26 Mechanism of absorption enhancer. Figure derived from Arbit E, et al[58] |
2017年, 诺和诺德便宣布FDA批准了索马鲁肽改善II型糖尿病患者的血糖控制。虽然索马鲁肽是长效的GLP-1激动剂, 但糖尿病患者仍需每周注射一次。为了提高患者的依从性, 诺和诺德很早就开始了口服索马鲁肽的研究。研究人员将索马鲁肽与吸收促进剂SNAC制成口服配方。SNAC与索马鲁肽结合使得其在胃部吸收, 而且溶解的SNAC在胃部形成局部相对较高的pH环境, 既可以增加索马鲁肽的溶解度, 在该环境下索马鲁肽受胃肽酶的影响很小, 又促进了索马鲁肽的吸收[59]。从图 27中可以看出, 口服索马鲁肽在胃部可以充分吸收并快速释放。2019年9月20日, 口服索马鲁肽被FDA批准用于结合饮食和运动以改善II型糖尿病患者的血糖控制。

|
图 27 Absorption and release of oral semaglutide. Figure derived from Knudsen LB, et al[59] |
含有疏水侧链的多肽往往水溶性较差, 而含有极性侧链的多肽水溶性相对较好。不同的多肽因其组成不同而具有不同的溶解性。有些临床使用的多肽药物常常含有芳香性氨基酸如苯丙氨酸、酪氨酸等, 但是这类含有芳香性氨基酸的多肽常常溶解性很差。胰高血糖素含有半数以上疏水性侧链, 且含有多个芳香性氨基酸, 因此, 其在水溶液中溶解性差。
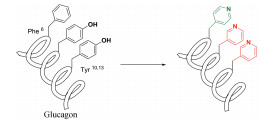
临床上通常使用胰高血糖素治疗急性低血糖。临床使用的胰高血糖素通常以冻干粉末的形式保存, 使用时需要用无菌酸性溶剂溶解, 但溶解时常常产生不溶性纤维[60, 61]。因此, 通过合适的修饰改造策略提高胰高血糖素的溶解性对胰高血糖素的临床使用有重要意义。天然的胰高血糖素85在PBS中的溶解度很小, Morz等[62]将胰高血糖素中的苯丙氨酸或酪氨酸替换为吡啶基丙氨酸3-pal或4-pal, 尤其是多个位点替换(表 5), 得到的肽在PBS溶液中的溶解性有了一定程度提高, 其中肽87和88, 在保持胰高血糖素活性的同时, 溶解度提高, 大于15 mg·mL-1。这也表明引入吡啶基团可以提高多肽分子的水溶性。Mayer等[63]也报道了利用吡啶基替换苯丙氨酸或酪氨酸中的苯环以提高多肽类降钙素基因相关肽受体拮抗剂的水溶性。
| 表 5 Replacing Phe/Tyr with pyridine motif to improve water-solubility of glucagon (Aib, aminoisobutyric acid) |
随着全球小分子药物研发的难度增加以及生物大分子药物研发速度的不断加快, 介于两者之间的多肽类药物也成为全球制药公司关注的焦点, 多肽药物的销售额也在逐年稳步上升。目前全球批准上市的多肽药物已超过80多种, 进入临床的多肽分子数量也在不断增加, 疾病领域涉及肿瘤、代谢性疾病、感染性疾病、免疫、心血管疾病以及泌尿生殖系统疾病等, 其中还有诸如甘精胰岛素和利拉鲁肽这种重磅炸弹级的多肽药物, 因此多肽药物的前景非常广阔。
多肽药物均衡了小分子药物和生物药的优点, 具有活性强、选择性好、安全性高, 不容易在体内蓄积、与其他药物相互作用少、代谢途径可预测等优点, 是一类理想的可用于开发成为药物的先导化合物。然而多肽药物本身也存在着半衰期短、血浆清除率高、不容易透过细胞膜、大多数药物不能口服, 通常需要注射给药, 患者依从性差以及生产成本较高等问题, 这些问题制约了多肽类药物的发展。采用多种化学修饰策略如末端结构修饰、拼接策略、环化策略、非天然氨基酸修饰、伪肽策略以及胆固醇修饰等可以提高肽类分子的活性; 除了上述部分方法可以提高肽的稳定性, 还可采用逆肽策略以及高级脂肪酸修饰、蛋白融合策略、聚乙二醇修饰等策略提高肽类分子的代谢稳定性; 采用引入卤素原子、去除极性侧链、手性策略、N-烷基化、高级脂肪酸修饰等策略可以改善肽类分子的渗透性。许多成功上市的多肽药物都用到了这些改造策略中的一种或几种。
目前, 通过对肽类分子的修饰和改造解决多肽药物的缺点, 仍然是最直接和有效的策略。熟悉了解多肽药物的基本改造策略, 对于多肽类药物的研究和开发具有重要意义。虽然多肽药物的发展仍然面临着一些挑战, 但随着未来药物化学改造策略的完善以及新型药物递送系统以及吸收促进剂的不断发展, 这些技术都将会应用到多肽药物的开发之中, 为多肽药物的开发提供更合理更丰富的思路。
| [1] |
Scognamiglio PL, Natale CD, Perretta G, et al. From peptides to small molecules:an intriguing but intricated way to new drugs[J]. Curr Med Chem, 2013, 20: 3803-3817. DOI:10.2174/09298673113209990184 |
| [2] |
Stoermer MJ, Chappell KJ, Liebscher S, et al. Potent cationic inhibitors of West Nile virus NS2B/NS3 protease with serum stability, cell permeability and antiviral activity[J]. J Med Chem, 2008, 51: 5714-5721. DOI:10.1021/jm800503y |
| [3] |
Yin Z, Patel SJ, Wang WL, et al. Peptide inhibitors of dengue virus NS3 protease. Part 2:SAR study of tetrapeptide aldehyde inhibitors[J]. Bioorg Med Chem Lett, 2006, 16: 40-43. DOI:10.1016/j.bmcl.2005.09.049 |
| [4] |
Schuller A, Yin Z, Brian Chia CS, et al. Tripeptide inhibitors of dengue and West Nile virus NS2B-NS3 protease[J]. Antiviral Res, 2011, 92: 96-101. DOI:10.1016/j.antiviral.2011.07.002 |
| [5] |
Leung D, Abbenante G, Fairlie DP. Protease inhibitors:current status and future prospects[J]. J Med Chem, 2000, 43: 305-341. DOI:10.1021/jm990412m |
| [6] |
Zhai Y, Zhao XS, Cui ZJ, et al. Cyanohydrin as an anchoring group for potent and selective inhibitors of enterovirus 713C protease[J]. J Med Chem, 2015, 58: 9414-9420. DOI:10.1021/acs.jmedchem.5b01013 |
| [7] |
Ma YY, Shang CY, Yang P, et al. 4-Iminooxazolidin-2-one as a bioisostere of the cyanohydrin moiety:inhibitors of enterovirus 713C protease[J]. J Med Chem, 2018, 61: 10333-10339. DOI:10.1021/acs.jmedchem.8b01335 |
| [8] |
Behnam MA, Nitsche C, Vechi SM, et al. C-terminal residue optimization and fragment merging:discovery of a potent peptide-hybrid inhibitor of dengue protease[J]. ACS Med Chem Lett, 2014, 5: 1037-1042. DOI:10.1021/ml500245v |
| [9] |
Driggers EM, Hale SP, Lee J, et al. The exploration of macrocycles for drug discovery——an underexploited structural class[J]. Nat Rev Drug Discov, 2008, 7: 608-624. DOI:10.1038/nrd2590 |
| [10] |
Xu SQ, Li H, Shao XX, et al. Critical effect of peptide cyclization on the potency of peptide inhibitors against Dengue virus NS2B-NS3 protease[J]. J Med Chem, 2012, 55: 6881-6887. DOI:10.1021/jm300655h |
| [11] |
Darnell JE. Transcription factors as targets for cancer therapy[J]. Nat Rev Cancer, 2002, 2: 740-749. DOI:10.1038/nrc906 |
| [12] |
Bromberg J, Darnell JE. The role of STATs in transcriptional control and their impact on cellular function[J]. Oncogene, 2000, 19: 2468-2473. DOI:10.1038/sj.onc.1203476 |
| [13] |
Yu H, Jove R. The STATs of cancer——new molecular targets come of age[J]. Nat Rev Cancer, 2004, 4: 97-105. DOI:10.1038/nrc1275 |
| [14] |
Gomez C, Bai LC, Zhang J, et al. Design, synthesis, and evaluation of peptidomimetics containing Freidinger lactams as STAT3 inhibitors[J]. Bioorg Med Chem Lett, 2009, 19: 1733-1736. DOI:10.1016/j.bmcl.2009.01.091 |
| [15] |
Chen JY, Bai LC, Bernard D, et al. Structure-based design of conformationally constrained, cell-permeable STAT3 inhibitors[J]. ACS Med Chem Lett, 2010, 1: 85-89. DOI:10.1021/ml100010j |
| [16] |
Patchett AA, Harris E, Tristram EW, et al. A new class of angiotensinconverting enzyme inhibitors[J]. Nature, 1980, 288: 280-283. DOI:10.1038/288280a0 |
| [17] |
Natesh R, Schwager SL, Evans HR, et al. Structural details on the binding of antihypertensive drugs captopril and enalaprilat to human testicular angiotensin I-converting enzyme[J]. Biochemistry, 2004, 43: 8718-8724. DOI:10.1021/bi049480n |
| [18] |
Chen CA, Sieburth SM, Glekas A, et al. Drug design with a new transition state analog of the hydrated carbonyl:silicon-based inhibitors of the HIV protease[J]. Chem Biol, 2001, 8: 1161-1166. DOI:10.1016/S1074-5521(01)00079-5 |
| [19] |
Mark WH. Francesco GS, Daniel HR. Synthetic and enzyme inhibition studies of pepstatin analogues containing hydroxyethylene and ketomethylene dipeptide isosteres[J]. J Med Chem, 1987, 30: 374-383. DOI:10.1021/jm00385a020 |
| [20] |
Blundell TL, Cooper J, Foundling SI, et al. On the rational design of renin inhibitors:X-ray studies of aspartic proteinases complexed with transition-state analogues[J]. Biochemistry, 1987, 26: 5585-5590. DOI:10.1021/bi00392a001 |
| [21] |
Sieburth SM, Chen CA. Silanediol protease inhibitors:from conception to validation[J]. Eur J Org Chem, 2006, 2006: 311-322. DOI:10.1002/ejoc.200500508 |
| [22] |
Wang C, Shi WG, Cai LF, et al. Artificial peptides conjugated with cholesterol and pocket-specific small molecules potently inhibit infection by laboratory-adapted and primary HIV-1 isolates and enfuvirtide-resistant HIV-1 strains[J]. J Antimicrob Chemother, 2014, 69: 1537-1545. DOI:10.1093/jac/dku010 |
| [23] |
Wang DX. Peptides and Drug Development (活性多肽与药物开发)[M]. Bejing: China Medical Science Press, 2008: 5.
|
| [24] |
Mustain WC, Rychahou PG, Evers BM. The role of neurotensin in physiologic and pathologic processes[J]. Curr Opin Endocrinol Diabetes Obes, 2011, 18: 75-82. DOI:10.1097/MED.0b013e3283419052 |
| [25] |
Seebach D, Lukaszuk A, Komisarska KP, et al. On the terminal homologation of physiologically active peptides as a means of increasing stability in human serum-neurotensin, opiorphin, B27-KK10 epitope, NPY[J]. Chem Biodiv, 2011, 8: 711-739. DOI:10.1002/cbdv.201100093 |
| [26] |
Sparr C, Purkayastha N, Yoshinari T, et al. Syntheses, receptor bindings, in vitro and in vivo stabilities and biodistributions of DOTA-neurotensin (8-13) derivatives containing bamino acid residues-a lesson about the importance of animal experiments[J]. Chem Biodiv, 2013, 10: 2101-2121. DOI:10.1002/cbdv.201300331 |
| [27] |
Aguilar ML, Purcell AW, Devi R, et al. β-Amino acid-containing hybrid peptides-new opportunities in peptidomimetics[J]. Org Biomol Chem, 2007, 5: 2884-2890. DOI:10.1039/b708507a |
| [28] |
Steer DL, Lew RA, Perlmutter P, et al. β-Amino acids:versatile peptidomimetics[J]. Curr Med Chem, 2002, 9: 811-822. DOI:10.2174/0929867024606759 |
| [29] |
Wang Y, Xing YH, Liu X, et al. A new class of highly potent and selective endomorphin-1 analogues containing alpha-methylene-beta-aminopropanoic acids (map)[J]. J Med Chem, 2012, 55: 6224-6236. DOI:10.1021/jm300664y |
| [30] |
Liu X, Wang Y, Xing YH, et al. Design, synthesis, and pharmacological characterization of novel endomorphin-1 analogues as extremely potent mu-opioid agonists[J]. J Med Chem, 2013, 56: 3102-3114. DOI:10.1021/jm400195y |
| [31] |
Wang DX. Peptides and Drug Development (活性多肽与药物开发)[M]. Beijng: China Medical Science Press, 2008: 573.
|
| [32] |
Gardoni F, Di Luca M. New targets for pharmacological intervention in the glutamatergic synapse[J]. Eur J Pharmacol, 2006, 545: 2-10. DOI:10.1016/j.ejphar.2006.06.022 |
| [33] |
Bach A, Eildal JN, Stuhr-Hansen N, et al. Cell-permeable and plasma-stable peptidomimetic inhibitors of the postsynaptic density-95/N-methyl-D-aspartate receptor interaction[J]. J Med Chem, 2011, 54: 1333-1346. DOI:10.1021/jm1013924 |
| [34] |
Taylor M, Moore S, Mayes J, et al. Development of a proteolytically stable retro-inverso peptide inhibitor of beta-amyloid oligomerization as a potential novel treatment for Alzheimer's disease[J]. Biochemistry, 2010, 49: 3261-3272. DOI:10.1021/bi100144m |
| [35] |
Hu XB, Nguyen KT, Jiang VC, et al. Macrocyclic inhibitors for peptide deformylase:a structure-activity relationship study of the ring size[J]. J Med Chem, 2004, 47: 4941-4949. DOI:10.1021/jm049592c |
| [36] |
Keri GR, Toth IN. Molecular Pathomechanisms and New Trends in Drug Research[M]. London and New York: Taylor & Francis, 2003.
|
| [37] |
Tyndall JD, Nall T, Fairlie DP. Proteases universally recognize beta strands in their active sites[J]. Chem Rev, 2005, 105: 973-999. DOI:10.1021/cr040669e |
| [38] |
Kawamoto SA, Coleska A, Ran X, et al. Design of triazole-stapled BCL9α-helical peptides to target the β-catenin/B-cell CLL/lymphoma 9(BCL9) protein-protein interaction[J]. J Med Chem, 2012, 55: 1137-1146. DOI:10.1021/jm201125d |
| [39] |
Rizzuti B, Bartucci R, Sportelli L, et al. Fatty acid binding into the highest affinity site of human serum albumin observed in molecular dynamics simulation[J]. Arch Biochem Biophys, 2015, 579: 18-25. DOI:10.1016/j.abb.2015.05.018 |
| [40] |
Liu ZG, Yu Z, Huang YY, et al. A novel stearic acid-modified hirudin peptidomimetic with improved pharmacokinetic properties and anticoagulant activity[J]. Sci Rep, 2015, 5: 14349-14360. DOI:10.1038/srep14349 |
| [41] |
Sjoholm A. Liraglutide therapy for type 2 diabetes:overcoming unmet needs[J]. Pharmaceuticals, 2010, 3: 764-781. DOI:10.3390/ph3030764 |
| [42] |
Lau J, Bloch P, Schaffer L, et al. Discovery of the once-weekly glucagon-like peptide-1(GLP-1) analogue semaglutide[J]. J Med Chem, 2015, 58: 7370-7380. DOI:10.1021/acs.jmedchem.5b00726 |
| [43] |
Iepsen EW, Torekov SS, Holst JJ. Liraglutide for type 2 diabetes and obesity:a 2015 update[J]. Expert Rev Cardiovasc Ther, 2015, 13: 753-767. DOI:10.1586/14779072.2015.1054810 |
| [44] |
Fujioka K, Sparre T, Sun LH, et al. Usability of the novel liraglutide 3.0 mg pen injector among overweight or obese adult patients with or without prior injection experience[J]. J Diabetes Sci Technol, 2015, 10: 164-174. |
| [45] |
Yang WQ, Xiao QQ, Wang D, et al. Evaluation of pharmacokinetic interactions between long-acting HIV-1 fusion inhibitor albuvirtide and lopinavir/ritonavir, in HIV-infected subjects, combined with clinical study and simulation results[J]. Xenobiotica, 2017, 47: 133-143. DOI:10.3109/00498254.2016.1166532 |
| [46] |
Beck A, Reichert JM. Therapeutic Fc-fusion proteins and peptides as successful alternatives to antibodies[J]. MAbs, 2011, 3: 415-416. DOI:10.4161/mabs.3.5.17334 |
| [47] |
Jimenez-Solem E, Rasmussen MH, Christensen M, et al. Dulaglutide, a long-acting GLP-1 analog fused with an Fc antibody fragment for the potential treatment of type 2 diabetes[J]. Curr Opin Mol Ther, 2010, 12: 790-797. |
| [48] |
O'Connor-Semmes RL, Lin J, Hodge RJ, et al. GSK2374697, a novel albumin-binding domain antibody (AlbudAb), extends systemic exposure of exendin-4:first study in humans-PK/PD and safety[J]. Clin Pharmacol Ther, 2014, 96: 704-712. DOI:10.1038/clpt.2014.187 |
| [49] |
Wang YS, Youngster S, Grace M, et al. Structural and biological characterization of pegylated recombinant interferon alpha-2b and its therapeutic implications[J]. Adv Drug Deliv Rev, 2002, 54: 547-570. DOI:10.1016/S0169-409X(02)00027-3 |
| [50] |
Hong Y, Zhou Y, Wang J, et al. Lead compound optimization strategy (4)——improving blood-brain barrier permeability through structural modification[J]. Acta Pharm Sin (药学学报), 2014, 49: 789-799. |
| [51] |
Liu HM, Liu XF, Yao JL, et al. Utilization of combined chemical modifications to enhance the blood-brain barrier permeability and pharmacological activity of endomorphin-1[J]. J Pharmacol Exp Ther, 2006, 319: 308-316. DOI:10.1124/jpet.106.106484 |
| [52] |
Perni RB, Britt SD, Court JC, et al. Inhibitors of hepatitis C virus NS3/4A protease 1. Non-charged tetrapeptide variants[J]. Bioorg Med Chem Lett, 2003, 13: 4059-4063. DOI:10.1016/j.bmcl.2003.08.050 |
| [53] |
Perni RB, Farmer LJ, Cottrell KM, et al. Inhibitors of hepatitis C virus NS3/4A protease. Part 3:P2 proline variants[J]. Bioorg Med Chem Lett, 2004, 14: 1939-1942. DOI:10.1016/j.bmcl.2004.01.078 |
| [54] |
Hu K, Geng H, Zhang QZ, et al. An in-tether chiral center modulates the helicity, cell permeability, and target binding affinity of a peptide[J]. Angew Chem Int Ed, 2016, 55: 8013-8017. DOI:10.1002/anie.201602806 |
| [55] |
White TR, Renzelman CM, Rand AC, et al. On-resin N-methylation of cyclic peptides for discovery of orally bioavailable scaffolds[J]. Nat Chem Biol, 2011, 7: 810-817. DOI:10.1038/nchembio.664 |
| [56] |
Beck JG, Chatterjee J, Laufer B, et al. Intestinal permeability of cyclic peptides:common key backbone motifs identified[J]. J Am Chem Soc, 2012, 134: 12125-12133. DOI:10.1021/ja303200d |
| [57] |
Hashizume M, Douen T, Murakami M, et al. Improvement of large intestinal absorption of insulin by chemical modification with palmitic acid in rats[J]. J Pharm Pharmacol, 1992, 44: 555-559. DOI:10.1111/j.2042-7158.1992.tb05463.x |
| [58] |
Arbit E, Goldberg M, Gomez-Orellana I, et al. Oral heparin:status review[J]. Thromb J, 2006, 4: 6-12. DOI:10.1186/1477-9560-4-6 |
| [59] |
Knudsen LB, Lau J. The discovery and development of liraglutide and semaglutide[J]. Front Endocrinol, 2019, 10: 155-186. DOI:10.3389/fendo.2019.00155 |
| [60] |
Onoue S, Ohshima K, Debari K, et al. Mishandling of the therapeutic peptide glucagon generates cytotoxic amyloidogenic fibrils[J]. Pharm Res, 2004, 21: 1274-1283. DOI:10.1023/B:PHAM.0000033016.36825.2c |
| [61] |
Pedersen JS, Dikov D, Flink JL, et al. The changing face of glucagon fibrillation:structural polymorphism and conformational imprinting[J]. J Mol Biol, 2006, 355: 501-523. DOI:10.1016/j.jmb.2005.09.100 |
| [62] |
Mroz PA, Perez-Tilve D, Liu F, et al. Pyridyl-alanine as a hydrophilic, aromatic element in peptide structural optimization[J]. J Med Chem, 2016, 59: 8061-8067. DOI:10.1021/acs.jmedchem.6b00840 |
| [63] |
Yan LZ, Johnson KW, Rothstein E, et al. Discovery of potent, cyclic calcitonin gene-related peptide receptor antagonists[J]. J Pept Sci, 2011, 17: 383-386. DOI:10.1002/psc.1358 |