 2020, Vol. 55
2020, Vol. 55

新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等多维性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
癫痫病是难治的中枢神经性疾病, 发病机制虽未完全清楚, 但已知谷氨酸AMPA受体亚型过分活跃是癫痫发作的重要原因。武田制药公司的抗癫痫药吡仑帕奈就是针对AMPA靶标研制的, 研制过程体现了以靶标为核心的理念和实施途径, 从发现先导物、结构优化、确定候选化合物, 到临床研究和上市, 紧密围绕着对靶标的活性和选择性以及化合物的成药性等内容, 成功地概念验证了结构优化的合理性。
(编者按)
谷氨酸是神经系统重要的兴奋性递质, 其生理效应是作用于两类受体蛋白:一是所谓代谢型谷氨酸受体亚型(mGluRs), 调节细胞的兴奋性和经过第二信使通路调节突触传导; 另一是离子移变型谷氨酸受体亚型(iGluRs), 为门控四聚型离子通道, 快速调节谷氨酸对突触的效应。iGluRs又基于对配体AMPA (1)、海人草酸(2)和NMDA (3)的选择性不同, 分成3种受体亚型, 这3个亚型的配体虽然都不是机体原有的天然配体, 但化学结构都是谷氨酸(4)模拟物。谷氨酸能神经传导的失调引起多种疾病, 例如癫痫、帕金森病、神经痛、卒中和记忆障碍等。选择性抑制AMPAs受体是治疗癫痫发作的重要途径, 但由于选择性作用不强或药代动力学性质不佳, 少有获得临床应用的药物(Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol, Toxicol, 2010, 50: 295-322)。

|
图 |
既往研究AMPA受体拮抗剂还没有上市的药物, 有代表性的化合物如竞争性拮抗剂替占帕奈(5, tezampanel)和非竞争性拮抗剂苄啶喹酮(6, piriqualone)等。5的结构中含有两个酸性基团和一个脂肪胺基团, 模拟谷氨酸竞争其结合位点; 6的结合位点不是谷氨酸结合部位, 所以化学结构中不含谷氨酸药效团特征(Welch WM, Ewing FE, Huang J, et al. Atropisomeric quinazolin-4-one derivatives are potent noncompetitive alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) receptor antagonists. Bioorg Med Chem Lett, 2001, 11: 177-181)。

|
图 |
用高通量筛选(HTS)方法在公司化合物库中搜寻先导化合物, 目标和标准是: ① AMPA受体非竞争性拮抗剂; ②剔除竞争性拮抗剂; ③剔除假阳性; ④化合物活性在μmol·L-1或以下水平。为此, 首先用AMPA诱导建立大鼠脑神经元细胞死亡模型, 评价化合物的阻断活性作为初筛, 评价对AMPA受体的拮抗活性。然后用3H-AMPA结合实验排除那些竞争性拮抗剂。再用AMPA诱导Ca2+流入, 测定抑制Ca2+离子50%进入细胞的化合物浓度(IC50), 确定拮抗性功能, 也据此排除假阳性化合物。通过HTS得到了噁二嗪酮为母核的化合物7 (IC50 9.17 μmol·L-1), 并作为先导化合物。
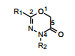
3 结构优化 3.1 对杂环2和4位苯基的变换用烷基或芳烷基对2和4位变换以探索构效关系, 合成的化合物及活性列于表 1。结果表明, 2位或4位任何一处变换为烷基或芳烷基都使活性降低, 提示这两个位置为苯环的必要性, 推测与噁二嗪酮环的共轭性是必要的。
| 表 1 Structure alteration at 2- and 4-position of the lead compound |
由于化合物7容易被代谢清除, 可能与噁二嗪酮环有关, 故用其他含氮六元杂环酮替换噁二嗪酮并保持2, 4-二苯基不变, 以优化活性和代谢稳定性, 合成的有代表性化合物的体外活性和清除率列于表 2。
| 表 2 Activity and metabolic stability of compounds with varied oxadiazinone moieties. a. H, M, and R stand for the in vitro clearance in men, mice and rats, respectively. The value smaller, the stability stronger |
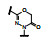
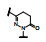
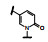
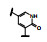
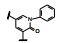
化合物13是二嗪酮为母核, 环上失去氧原子使活性降低; 14和15是吡啶酮化合物, 活性也低于先导物13, 化合物15的氮原子上有活泼氢, 代谢稳定性差。16是用苯环替换了氢, 活性和稳定性不仅优于14和15, 而且优于先导物7, 从而优化出三苯基吡啶酮的结构。
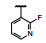
4 三苯基的变换 4.1 确定母核位置的优化基团以1, 3, 5-三苯基吡啶酮为新的起点, 用吡啶环分别替换不同位置的苯环以优化活性, 表 3列出了有代表性的化合物。化合物17和18抑制AMPA受体阻止Ca2+流入的活性强于16, 提示母核吡啶酮的5位(Ar1)连接2′-吡啶基有利于活性, 为此, 固定C5位2′-吡啶基作深入优化。
| 表 3 Activity of the compounds with pyridine instead of phenyl ring |

C3连接的芳环邻近于吡啶酮的羰基, 环上的取代基电性和立体性可能对羰基与受体的结合有重要影响, 为此, 固定N1-苯基和C5为2′-吡啶基, 变换C3的连接基, 化合物的结构与活性列于表 4。
| 表 4 Activity of compounds with alteration in C3 linkers |
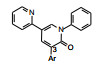
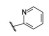
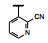
表 4中有代表性的化合物并非批量一次合成的, 而是首先选取了氰基取代苯, 氰基是拉电子基团, 也有较大的体积, 连接在不同位置对母核吡啶酮的电性分布以及C3-苯环同母核环的扭转程度是不同的, 从而可能影响活性。结果表明2′-氰苯基化合物(19)的活性强于无取代的17, 氰基移至间位(20)或对位(21), 活性依次下降, 提示2′-取代的位阻效应使C3环-吡啶环形成有利于结合的两面角。这样, 考察其他原子或基团的效应都连接在2′-位。化合物22~25的2′-不同取代的化合物有较高的活性, 但都低于19。用3-(2′-取代)吡啶基替换苯基, 化合物26~28活性弱于相应的苯基化合物。至此, 19是活性优化最强的化合物。
4.3 以化合物18为起点考察C3的芳环效应化合物18为N1, C5二吡啶取代物, 也是活性较高的化合物, 因而以18为出发点考察C3的基团变换对基团的影响。表 5列出了化合物的活性。
| 表 5 Activity of compounds with varied C3-aromatic moieties |

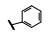
表 5中的化合物29和32虽然活性低于19, 但仍证明2′-氰基取代是优选的取代基。不过也说明N1为苯环优于吡啶环取代。
4.4 固定C3为2′-氰苯基、N1为苯基, 优化C5取代基药物化学中优化多位置的化合物时, 往往变换一个位置而固定其余结构, 这样优化局部结构所得到的结果并非固定不变的, 当变换另一位置的结构时, 原先的优化结论未必适用, 这就是反复优化的原因。下面就是C3和N1分别固定为氰苯基和苯基, 探索C5的最佳取代。表 6列出了化合物的结构和活性。
| 表 6 Structure and activity of compounds with varied C5-moieties |
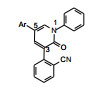
表 6的数据表明C5变换成取代苯基或噻吩基的活性都低于2′-吡啶基, 变换吡啶基的连接位置也使活性降低, 因而19仍是体外活性最强的化合物。
4.5 固定C3为2′-氰苯基、C5为2′-吡啶基, 优化N1取代基至此, 将C3固定为2′-氰苯基、C5为2′-吡啶基, 再优化N1取代基。表 7列出了化合物活性。结果表明, 变换吡啶基或环上引入不同取代基, 活性都弱于19。
| 表 7 Structures and activity of compounds with varied N1-moieties |
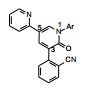
在以吡啶酮为母核的N1, C3, C5-三芳基取代的优化中出现的一些高活性化合物, 测定了对微粒体的代谢稳定性和灌胃小鼠抑制AMPA引起惊厥的最低有效剂量, 表 8列出了评价结果。表明化合物19、26、28和34对小鼠有较强的抗惊厥活性(体内), 对人、小鼠和大鼠肝微粒体有良好的代谢稳定性(体外), 其中化合物19尤其突出。
| 表 8 In vitro and in vivo metabolic stability of compounds with high activity. a H, M, and R stand for the in vitro clearance in men, mise and rats, respectively. b Minimum effect dose |
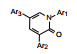



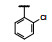
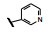
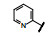
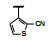


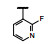
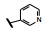
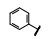
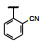

|
图 |
化合物19灌胃大鼠(1 mg·kg-1)表明有良好的药代动力学性质, 口服生物利用度64.3%, Cmax 0.17 μg·mL-1, 半衰期t1/2 2.37 h。还评价了化合物19在小鼠和大鼠脑与血浆中的浓度比值, 分别为1.06和1.14。小鼠脑脊髓液中19的浓度与血浆中游离浓度的比值为1.14。这些数值表明化合物19作为中枢神经系统药物适宜穿越血脑屏障。此外, 还评价了对63种酶、受体和离子通道的作用, 提示没有脱靶作用, 有较高的选择性作用, 遂确定为候选化合物进入临床前研究, 命名为吡仑帕奈(perampanel) (Shigeki Hibi S, Ueno K, Nagato S, et al. Discovery of 2‑(2-oxo-1-phenyl-5-pyridin-2-yl-1, 2-dihydropyridin-3-yl)benzonitrile (perampanel): a novel, noncompetitive α‑amino-3-hydroxy-5-methyl-4-isoxazolepropanoic acid (AMPA) receptor antagonist. J Med Chem, 2012, 55: 10584-10600), 经三期临床研究表明是作用于AMPA受体非竞争性的拮抗剂, 用于口服治疗部分发作型的癫痫病, 美国FDA于2012年批准上市。