 2019, Vol. 54
2019, Vol. 54

新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等多维性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
帕布昔利布是针对驱动细胞分裂的靶标研制的首创性药物, 难度在于在众多的周期蛋白依赖性激酶中仅针对CDK4选择性活性的问题。从苗头化合物到成功上市, 骨架结构虽然没有改变, 但外周的基团“转着圈儿地”变换, 反复地优化, 甚至在芳环上改换一个原子(苯环改成吡啶环), 其他各个位置重新优化, 其中不乏有高活性的化合物, 但确定候选化合物则要综合所有的成药性质于结构一身, 药物化学、分子模拟和构效分析的经验、智慧和决策是注定的前提。
(编者按)
正常细胞的增殖是通过严格的细胞周期性变化而完成的, 从一次分裂完成到下一次分裂结束称作细胞周期(cell cycle), 包含4个阶段: ①合成RNA和蛋白质, 称作合成前期或G1期, 为下一步的DNA复制做好物质和能量准备; ②进行DNA合成的S期, 同时还合成组蛋白和DNA复制所需的各种酶; ③细胞分裂的G2期即DNA合成后期, 此时DNA停止合成, 为下阶段的分裂要合成大量的RNA和蛋白质; ④细胞分裂的M期, 通过有丝分裂由一个母细胞分裂成两个子细胞。[G1→S→G2→M]构成一个细胞周期。
1.2 周期蛋白依赖性激酶与细胞周期细胞周期的运行是靠周期蛋白(cyclin)和周期蛋白依赖性激酶(cyclin-dependent kinases, CDK)的协同调控实现的。人体有21种CDK, 是一组丝/苏氨酸蛋白激酶; 还有16种cyclin, 是一类同源蛋白。不同的CDK与不同的cyclin结合形成的复合物, 通过磷酸化, 实现对细胞周期不同时相的推进和转化。直接与细胞周期4个阶段相关的CDK只有7个(CDK1~4, 6, 10, 11), 其余的功能是间接调控。图 1是不同CDK/cyclin在细胞周期的作用(Sánchez-Martínez C, Gelbert LM, Lallena MJ, et al. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs. Bioorg Med Chem Lett, 2015, 25: 3420-3435)。

|
Figure 1 The role of various CDK/cyclins in cell cycle |
细胞周期蛋白依赖性激酶和周期蛋白的发现以及阐明它们驱动细胞分裂的功能是生物学上的重要发现, 为此获得了2001年诺贝尔生理学或医学奖(获奖者是美国的Hartwell、英国的Hunt和Nurse)。
以抑制细胞增殖为目标最先研究的药物靶标是CDK2, 但候选物的临床试验未呈现预期疗效。后来的实验表明, 用siRNA抑制CDK2并不能抑制骨肉瘤和Rb阴性的脑瘤的生长, 说明CDK2不是药物治疗肿瘤的有效靶标(Tetsu O, McCormick F. Proliferation of cancer cells despite CDK2 inhibition. Cancer Cell, 2003, 2: 233-245)。
1.3 CDK4/D和Rb抑癌基因在细胞核内存在有成视网膜细胞瘤基因(Rb), 表达蛋白Rb为肿瘤抑制素, 在CDK4/cyclin D的作用下被磷酸化成p-Rb, 由此导致结合在其上的E2F释放, E2F是转录因子, 促进S期所必需的周期蛋白和CDK蛋白的转录, 使细胞从G1期进入S期。由G1过渡到S期的限制点是由CDK4/cyclinD和CDK6/cyclinD (简称CDK4/D, CDK6/D)调控的, 已成为当前肿瘤学研究的重要靶标。肿瘤细胞的CDK4/Rb通路异常, 例如周期蛋白D的过高表达、CDK4突变、p-Rb的突变或缺失等, 肿瘤病毒编码的癌蛋白与癌细胞的p-Rb结合, 使其失去结合E2F的能力, 导致细胞不断分裂而恶性增殖。因而选择性CDK4抑制剂可恢复正常细胞的功能, 用于治疗癌症和细胞生长失控的疾病。
2 前期抑制剂的研究 2.1 研发目标CDK/cyclin是调控细胞增殖的重要枢纽, 泛CDK抑制剂的无选择性会造成正常细胞的损伤, 脱靶作用产生不良反应。多数肿瘤细胞的CDK4 (6)/ cyclin通路高度活跃, 因而选择性作用于CDK4 (6)/ cyclin应是抗肿瘤药物的研制目标, 但要避免对其他CDK的抑制作用。
2.2 苗头化合物的由来研发CDK/cyclin抑制剂始于20世纪90年代。Parke-Davis公司将研发酪氨酸激酶(如EGF、FGF、PDGF和c-Src)抑制剂合成的化合物进行筛选, 经生化普筛、分析CDK2-cyclin复合物晶体结构和分子模拟等方法, 发现骨架为嘧啶并吡啶酮的化合物1对CDK4/D有一定的选择性抑制活性。

|
分子模拟和构效关系(有效和无效分子的结构差异)分析表明, 这类对酪氨酸激酶有活性的化合物都在6位有芳环存在, 6位苯基进入深部体积较大的疏水腔中(邻近于ATP结合位点), 而同源模建的CDK4结构中相应疏水腔很小, 而且被体积大的氨基酸残基遮盖, 既然6位芳基有碍于结合, 所以苗头化合物1的6位没有苯基取代。
2.3 2位苯胺基的变换以化合物1为起始物, 固定N8的乙基不变, 变换C2连接的苯胺片段, 合成的化合物和抑制CDK4/D活性列于表 1。
| Table 1 Structure and activity of the compounds with varied C2-linking fragments |


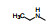
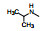
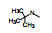

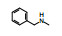


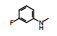
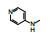
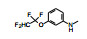
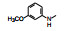




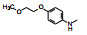
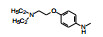
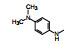


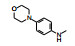
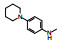
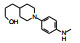
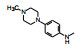
表 1的构效关系分析如下: ①用小的烷基(2~4)或环己基(5)置换苯基都使活性下降, 例如5的活性降低了5倍。②苄胺基(6)和N-甲基(7)都失去活性(IC50 > 40 μmol·L-1), 是在预料之中的, 因为分子模拟显示1的NH与CDK2的Leu-83 (CDK4为Val-96)形成氢键。6的NH位置变动并过于碱性, 7的NCH3无氢, 失去氢键给体的能力。③苯环上引入拉电子基团4-F或3-F (化合物8和9)使活性下降。④苯环上烷氧基取代, 活性取决于在环上的位置和烷基大小。例如4-甲氧基(13)活性与1相当, 而3-甲氧基(12)没有活性。长链的烷氧基(17)活性减弱, 但含氮的长链(18)的活性较强, 该构效关系说明结合的复杂性。⑤ 4位含氮的片段有利于活性, 特别是成环后如4-N-甲基哌嗪(25)比1的活性强6倍。
优化结论:苯环4'位连接哌嗪环有利。
2.4 N8的乙基变换固定C2为苯胺基(无哌嗪基是为了合成方便), 变换N8的烷基, 合成的化合物活性列于表 2。
| Table 2 Structure and activity of the compounds with varied N8-linking fragments |
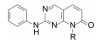





表 2的构效关系显示: ① N8连接含氧原子的烷基链活性都很弱, 例如化合物40~44, 说明极性原子不利于该位置的结合。② N8连接较大的亲脂性基团有利于活性, 例如2-降莰烷基(36)的活性很高, 环烷或大的链烷基活性强于芳烃基(如34强于37)。
优化结论: N8宜连接较大的环烷基。
2.5 含哌嗪环的N8基团取代在C2的苯胺基4位连接碱性侧链可提高抑制CDK4/D的活性。固定4'-哌嗪基不变, 变换N8的片段, 化合物活性列于表 3中。
| Table 3 Structure and activity of the piperazine-containing compounds with varied N8-linking fragments |




化合物53综合了上述两个优化因素, 是活性最强的化合物, 强于1大约150倍, 显示出C2与N8的优化的叠加效果。
2.6 对CDK4/D的选择性对高活性的化合物评价抑制CDK4/D的选择性作用, 表 4列出了对CDK1/B、CDK2/A、CDK2/E和FGFR (酪氨酸激酶)的活性, 对这几个激酶的高活性意味着不利的脱靶作用。
| Table 4 Selective inhibition of typical compounds on CDK4/D |
表 4中化合物对CDK亚型和FGFR活性的总趋势是, 对CDK4/D活性高的化合物对其他亚型的活性也高, 例如化合物53对CDK4/D的活性是49的8倍, 对其他亚型的作用53也比49强。53抑制CDK4/D的活性3倍强于CDK2/A, 49强于CDK2/A大约2倍。其余的化合物则对CDK2/A的活性显著强于CDK4/D。这些说明, 通过变换C2或N8的基团可调节对CDK的选择性作用(Barvian M, Boschelli DH, Cossrow J, et al. Pyrido[2, 3-d]pyrimidin-7-one inhibitors of cyclin-dependent kinases. J Med Chem, 2000, 43: 4606-4616)。
3 抑制剂与CDK4/D结合的特征由于只有CDK2/A的晶体结构, 对CDK4/D的结合模式是靠同源模建后作分子模拟实现的。化合物49与CDK2/A的晶体结构的X-射线衍射分析表明, N3与Leu-83骨架的NH形成氢键, C2侧链上的NH作为氢键给体也与Leu-83形成氢键, 这两组氢键在ATP的结合模式或其他结构类型的抑制剂是没有的, 它们虽然都占据了ATP的结合位点, 但不与Leu-83形成氢键, 而是分子中的NH与Glu-81的羰基形成氢键, 但49不存在这种结合。图 2是化合物49与CDK2/A复合物的晶体结构图。在母核的5, 6位处有Phe80 (相应于CDK4/D为Phe93)的覆盖, 对5或6位存在的较大的取代基构成位阻作用。

|
Figure 2 Crystallographic diagram of the complex between compound 49 and CDK2/A |
分析化合物49与嘌呤类抑制剂[如puralanol A (54)]在CDK2/A活性部位的结合模式(无图显示), 发现54的N9位所连接的异丙基与49的C5所处的位置相当, 推测49的C5虽接近于Phe80 (于CDK4/D为Phe93), 但仍可能有一定的结合空间, 因而考察嘧啶并吡啶酮的5位取代对活性的影响, 由于分子模拟表明空间不大, 故取代基限定于甲基取代。表 5列出了化合物的结构和抑制CDK4/D的活性。

|
表 5的构效关系表明, C5引入甲基的化合物(59~63)抑制CDK4/D的活性弱于无取代的相应化合物(51, 55~58), 提示这个位置的甲基取代使活性减弱。与当初puralanol A可容许异丙基的预示不符, 至少不能把puralanol A与CDK2/A的结合模式套用于嘧啶并吡啶酮与CDK4/D的结合。然而重要的是, 5-甲基化合物对其他CDK的活性显著降低, 提高了抑制CDK4/D的选择性作用, 降低对CDK1和CDK2的脱靶作用。权衡得失, 5-甲基取代是有利的, 因为分子设计旨在成功药物, 不在于单纯追求高活性化合物。
| Table 5 Structure and activity of the compounds with varied C5 substituents |

化合物63对CDK4/D具有高活性和高选择性, 进而以63为新的起点, 考察变换C5 (与甲基尺寸相近的基团)和N8的取代基, 优化活性与选择性, 表 6列出了有代表性的化合物。
| Table 6 Structure and activity of the compounds with varied C5 and N8 groups |
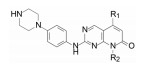

表 6的数据显示R1增大为乙基(64), 活性降低50倍, 换成三氟甲基(65)也显著减弱, 推测是由于体积和亲电性加大的缘故。所以C5以甲基为优化基团。66和67的N8基团分别为异丙基和异戊基, 活性也降低, 如前所述, N8连接的片段处于ATP的核糖位置, 已证明可容许大基团如降莰烷基(化合物36和52)的结合, 仍保持强活性。
优化结论: C5为甲基, N8为环戊基。
4.3 C2片段上含氮杂环的优化固定C5为甲基, N8为环戊基, 考察C2连接的末端碱性基团对活性的影响, 表 7列出了化合物的活性和选择性。
| Table 7 Structure and activity of the compounds with piperazine alteration |
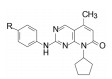
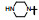

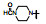

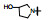
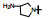

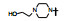
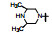
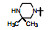
表 7的构效关系表明, 以化合物63为基准, 哌嗪的N4'被酰化(68~70)活性显著下降, 3'-羟基四氢吡咯(71)的活性也降低10倍多, 但3'-氨基四氢吡咯(72)、二氮杂环庚烷(73)以及3', 3'-偕二甲基哌嗪(76)的活性与63相近, 且保持高选择性。所以, 含有可离解的氮原子是非常必要的, 可能与酶Asp99发生氢键或盐键结合作用, 并非只起助溶性基团的作用。化合物75是3', 5'-二甲基哌嗪, 活性降低10倍, 可能因占据空间太大, 不能被结合腔包容。
5 6位取代基对活性的影响化合物49与CDK2/A的晶体结构(图 2)显示, 母核的5位和6位靠近Phe80, 不宜有取代基, 但CDK4/D与CDK2/A未必相同, 上一节证实了5-甲基取代基本保持了对CDK4/D的活性, 重要的是降低了对其他CDK的作用。这也提示人们对6位的取代也应做精准的研究和试错(trail and error)性探索。以化合物63为基准, 探索6位的变化对活性的影响, 化合物列于表 8。
| Table 8 Structure and activity of the compounds with varied 6-position substituent |

分析表 8化合物的构效关系如下: ① 5, 6-二甲基(77)对CDK4/D的活性显著比6位无取代的(63)低10倍, 但保持了高选择性。当6位被乙基取代, 活性与63相当, 但选择性降低到61倍。可见6位取代基的性质对活性和选择性是非常敏感的, 反映了CDK4抑制剂的活性具有悬崖式的变化。② 6位分别用氟氯溴碘原子取代对活性和选择性产生有趣的变化:随着原子尺寸加大对CDK4/D的活性增加, 例如6-氟(79)的IC50 = 0.03 μmol·L-1, 6-碘代(82)为0.005 μmol·L-1。但增大原子尺寸又使对CDK4/D选择性降低了。所以, 活性强度与选择性之间存在有微妙的平衡, 需要精细的结构调整, 实现高活性与高选择性的共存或叠加。③为了模拟卤素拉电子效应以提高对CDK4/D的活性且不降低选择性作用, 合成了6-乙酰基(83)、羧基(84)和酯基(85和86)化合物。6-乙酰基活性最强, 但选择性不够强, 对CDK2/A的活性只差100倍。6-乙酯基则是活性和选择性都高的化合物。
对化合物83、85和86评价了抑制人结肠癌(HCT116)和人乳腺癌(MDA-MB-435)的细胞活性, IC50的范围是0.032~1.35 μmol·L-1。进而用移植人乳腺癌裸鼠评价化合物83、85和86的抗癌作用, 结果表明化合物83显示有抗癌效果, 而两个酯化合物未呈现活性, 是由于在体内水解成游离酸的缘故(细胞培养未见水解) (VanderWel SN, Harvey PJ, McNamara DJ, et al. Pyrido[2, 3-d]pyrimidin-7-ones as specific inhibitors of cyclin-dependent kinase 4. J Med Chem, 2005, 48: 2371-2387)。
6 C2连接的2'-氨基吡啶系列的优化初始研究CDK4/D抑制剂时, 发现将化合物33的C2-苯胺基用电子等排体2'-氨基吡啶置换的化合物(87), 其选择性作用显著高于33, 随后在88和89一对化合物之间也呈现吡啶等排体的选择性强于苯基, 表 9列出了这两对化合物的活性和选择性数据。
| Table 9 Comparison of the activity and selectivity between C2-anilino and 2'-aminopyridyl compounds |
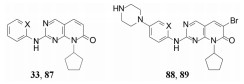
虽然吡啶置换苯环后抑制CDK4/D活性有所减弱, 但选择性显著优胜于苯系列, 为此对吡啶系列作新一轮的优化, 按照前面叙述的内容实施操作。
7.1 N8位置的优化根据前述在N8位置的优化片段, 合成了有限的化合物列于表 10中, 构效关系表明, 在C2新的取代基的条件下, N8-环戊基是活性和选择性最佳的结构片段, 例如化合物92对CDK4/D选择性强于93, 从而优化其他位置的化合物都固定N8为环戊基。
| Table 10 Structure and activity of the compounds with varied N8 substituents |


前述的C6-溴代化合物(89)显示有良好的选择性, 但因原子量大和极性小, 需进行优化, 合成的化合物列于表 11。
| Table 11 Structure and activity of the compounds with varied C6 substituents |

表 11的构效关系为: ① C6的各种取代仍然保持对CDK4/D的选择性作用。②简单的极性基团如氨基(95)或非极性基团如乙基(97)直至长链的甲氧乙氧甲基(101)都有较高的活性和选择性。但异丁氧基的活性显著下降, 可能是末端体积过大的缘故。③乙酰基(105)和羧酸甲酯(106)的活性都较弱, 可能是由于C5没有甲基取代的缘故。
7.3 C5-甲基取代的重要性基于前述的苯胺系列的构效关系, 在吡啶系列中考察C5的甲基取代对活性的影响, 将C2连接的末端固定为哌嗪基, C6选取活性较高的溴、乙酰基和羧酸甲酯, 合成的化合物列于表 12。
| Table 12 Structure and activity of the compounds with varied C5, C6 and C2 groups |

表 12的基团变换对活性的影响表明, C5被甲基取代、C6被溴或极性基团取代、C2连接的吡啶环上有哌嗪基是高活性和选择性的三要素, 缺一不可(6-溴代化合物无甲基化合物89的活性强于有甲基的110, 但其选择性差)。由于酯基在体内易被水解, 故只进一步比较6-溴和6-乙酰基的活性贡献, 特别要考察C2末端的碱性片段的性质对活性的影响。
7.4 C2-氨基吡啶系列的碱性基团的优化优化至此, 化合物113是活性和选择性最高的化合物, 基团的配置与苯胺系列的高活性化合物83是相同的。然而氨基吡啶系列并没有对C2的末端碱基进行优化。由于高活性化合物多出现在5-甲基-6-溴和5-甲基-6-乙酰基化合物中, 因而优化碱基固定在这两个亚系列中。表 13列出了化合物的抑酶活性(CDK4/D)、选择性(CDK2/A)、和人乳腺癌细胞(MDA-MB435)的活性。
| Table 13 Structure and activity of the compounds with optimized alkali groups in the series of aminopyridines |


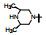

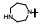
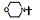

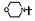


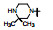

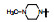

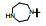




表 13的构效关系显示, 6-溴和6-乙酰基两个亚系列基本保持了抑制CDK4/D的活性和高选择性(对CDK2/A的IC50 > 5 μmol·L-1), 6-乙酰基化合物的活性强度普遍高于6-溴化合物。在6-乙酰化合物中, 哌嗪基(113)、氨基四氢吡咯基(127)、二氮杂环庚烷基(128)和吗啉基等都呈现高活性和选择性。对乳腺癌细胞也有强效的活性(Toogood PL, Harvey PJ, Repine JT, et al. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J Med Chem, 2005, 48: 2388-2406)。
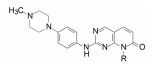
8 候选物的确定和帕布昔利布的批注上市在高活性和选择性的化合物中, 113显示出优于其他的化合物在于对CDK6与CDK4有相同的活性, CDK6在一些肿瘤生长中是高表达的激酶, 113对其他36个激酶未显示活性, 提示高选择性地抑制Rb阳性细胞于G1期。大鼠的药代动力学表明口服生物利用度F = 56%, 清除率CL = 37.5 mL·min-1·kg-1, 半衰期t1/2 = 2.1 h, 同时也有良好的物理化学性质。辉瑞公司确定113为候选化合物, 定名为帕布昔利布(palbociclib), 经三期临床研究于2015年FDA批准上市, 治疗雌受体阳性、HER2阴性的乳腺癌。

|
这是针对驱动细胞分裂的靶标研制的首创性抗癌药物, 难度在于从众多的周期蛋白依赖性激酶只针对CDK4 (和6)。从苗头化合物1到上市的药物帕布昔利布(113), 骨架结构—嘧啶并吡啶酮没有改变, 但外周的基团“转着圈儿地”变换, 而且是反复地修饰, 甚至在将C2连接的苯环改成吡啶环后, 再一次地逐个位置的优化, 因为更换一个原子可能对活性或选择性带来悬崖式的变化。图 3是将研发帕布昔利布的重要节点用化学结构加以说明。

|
Figure 3 Scheme of the optimization of palbociclib |