 2019, Vol. 54
2019, Vol. 54

新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等多维性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
药物治疗肺动脉高压病可针对不同的靶标和环节。利奥西呱是以激活可溶性鸟苷酸环化酶为靶标的首创性药物。基于生理机制研发药物的重要前提是靶标的可药性, 因而揭示对靶标的作用与临床效果的因果关联性贯穿于首创药物的研发始终, 概念验证是反复进行的。利奥西呱的研制过程中用酶、器官、多种动物的病理模型评价化合物的活性和选择性, 以保障临床的有效性。结构优化也是在药效、药代和选择性等多维空间中展开的。
(编者按)
肺动脉高压(plumonary artery hypertension, PAH)指肺动脉压力升高, 超过一定界值而发生的血流动力学和病理生理状态, 可导致右心肥大和衰竭, 是一种致残率和病死率很高的疾病。患者内皮素浓度明显增高, 尤其是肺动脉血中内皮素浓度显著高于右心室和股动脉, 由于肺动脉平滑肌细胞的异常收缩和增殖, 导致肺动脉壁重塑(平滑肌细胞增生、凋亡、炎症和纤维化等复合作用引起血管结构的病理变化), 引起血管阻力和压力增大等病变。
2 药物治疗的环节为了阻断和缓解肺动脉壁的重塑, 药物应减轻患者肺动脉血管内壁的纤维化, 阻止内皮细胞的增生以及平滑肌肥大和增生等。就药物作用靶标而言, 主要聚焦于3个方面, 即①内皮素受体拮抗剂; ②前列环素(PGI2)受体激动剂; ③一氧化氮(NO)受体激动剂。
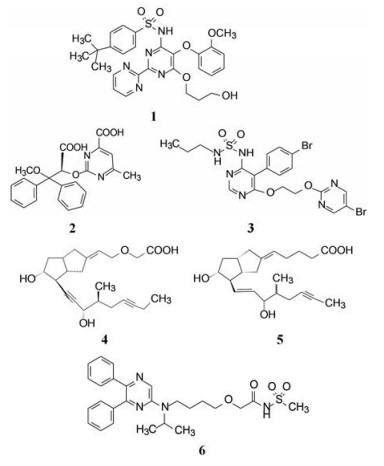
2.1 内皮素受体拮抗剂内皮素(ET-1)是引起肺动脉重塑和PAH的主要祸害, 阻断ET-1对受体ETA和ETB的结合, 上市药物已有波生坦(1, bosentan)、安倍生坦(2, ambrisentan)和马西替坦(3, macitentan)等, 都是口服用药。
2.2 前列环素受体激动剂前列环素(PGI2)是内源性激素, 主要产生于血管内皮细胞。PGI2具有多种生理功能, 可强效舒张血管, 抑制血管平滑肌细胞分化、增殖和迁移以及抑制血小板聚集。上市的PGI2受体激动剂药物有伊洛前列素(4, iloprost)和西卡前列素(5, cicaprost)以及赛乐西帕(6, selexipag)。
|
健康人体内的一氧化氮(NO)的功能是舒张血管和抑制平滑肌细胞增生, 机制是通过激活可溶性鸟苷酸环化酶(sGC)提高生成环鸟苷酸(cGMP)水平。然而NO释放剂如硝酸酯对PAH患者无效, 是由于微弱的肺血管舒张作用被强大的扩张外周血管抵消, 不良反应超过主作用。因而需通过激活可溶性鸟苷酸环化酶提高患者cGMP水平, 即开cGMP之源。另一条途径是节cGMP之流。例如抑制磷酸二酯酶5 (PDE5)活性, 保护和延缓cGMP免于水解成GMP, 维持对肺动脉的舒张。所以, 治疗男性勃起障碍的药物西地那非(sildenafil)和他达拉非(tadalafil)也用于治疗PAH。
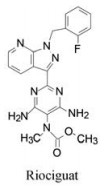
本文叙述激活可溶性鸟苷酸环化酶药物利奥西呱(7, riociguat)的研发过程。

|
1994年台湾大学Ko等发现化合物8 (YC-1)可抑制多种炎症介质引起家兔的血小板聚集, 并证明是激活了血小板的可溶性鸟苷酸环化酶(sGC), 提高了cGMP水平, 因而8作为激活sGC的苗头化合物, 开辟了研制舒张血管防止血小板聚集药物新方向(Ko FN, Wu CC, Kuo SC, et al. YC-1, a novel activator of platelet guanylate cyclase. Blood, 1994, 84: 4226-4233)。
评价受试化合物的体外活性用两种方法。一是家兔主动脉环模型, 用一定浓度的苯肾上腺素处理主动脉环引起收缩, 测定并计算达到设定舒张程度的化合物浓度IC50。另一是用重组的可溶性鸟苷酸环化酶在NO存在下产生cGMP的最低有效浓度(MEC)来评价化合物活性。化合物的体内活性是灌胃大鼠测定麻醉状态下的扩张血管、降低血压的作用。
4 结构优化Bayer公司以化合物8为先导化合物。为进行结构修饰, 将8分成3部分分别优化, 即苄基, 苯并吡唑和羟甲基呋喃。
4.1 苄基是必要的将连接苯并吡唑与苯基的亚甲基变换成-COO-、-CO-或单键直接连接, 合成的化合物都失去或降低了失活, 提示母核上苄基取代的必要性, 因而以后的结构修饰不改变这个连接片段。在苯环上做不同位置的基团取代, 或用杂环置换苯环, 合成的有代表性的化合物列于表 1。
| Table 1 Structure and activity of the compounds with different N-substituents |
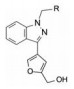
表 1的构效关系提示, 苯环的2-位氟代化合物的活性强于3或4位氟代, 2-CN同样强于3-CN。其他卤素取代均使活性降低(数据未列)。虽然5-嘧啶取代的化合物活性最强, 但溶解性差, 而且难以合成, 所以, 选取了化合物9作为继续优化的起点。
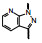
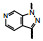
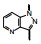
4.2 母核苯并吡唑的变换母核苯并吡唑用其他双环进行了变换, 合成的化合物列于表 2, 结果显示吡唑并吡啶环连接的化合物19活性最强, 而且氮原子的位置很重要, 20~22的化合物活性弱, 而且多一个氮原子的吡唑并嘧啶化合物23活性也差。所以, 19为优选的化合物。
| Table 2 Activity of the compounds with varied benzopyrrazole rings. aValue before parenthesis is concentration, inside is inhibition percentage |
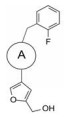
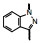


将母核固定为吡唑并吡啶, N1连接的是2-氟代苯甲基, 下一步考察变换呋喃环对活性的影响, 表 3列出了化合物的结构与活性。
| Table 3 Structure and activity of the compounds with various heterocycles at C3 position |
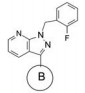
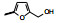

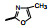
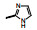
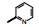
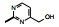
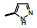
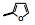
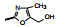
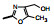
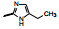
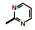

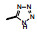
表 3的数据表明, 呋喃环上去除羟乙基或换成磺酰胺基都使活性降低, 用噁唑、咪唑或吡唑环替换呋喃, 活性基本不变或略低, 2-嘧啶基化合物32活性略高, 虽然加入羟甲基活性未见提高, 但4-氨基嘧啶(34)活性显著增强。
4.4 氨基嘧啶环上取代基变换母核上C3连接的4-氨基嘧啶环是优选的片段, 进而对环上C5做取代基修饰, 合成的化合物列于表 4。
| Table 4 Activity of the compounds with varied substituents at pyrimidine ring |
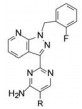
表 4的构效关系可总结如下: ①在2-氨基嘧啶环的5位做不同的取代, 除正己基、氰庚基和氨甲基外, IC50都在1 μmol·L-1以下, 例如烷基取代C2~C5的活性变化不大, 说明氨基嘧啶环的C5-基团变动影响不大; ②立体位阻较大或亲脂性强的基团使活性减弱; ③含有极性基团(同样链长)的化合物活性较强, 例如吗啉基比硫代吗啉的活性强一倍。但氨甲基化合物64活性很弱, 或许是部分离解的缘故。
4.5 里程碑式化合物45表 4中有若干个活性(IC50)低于300 nmol·L-1的化合物, 但拜耳公司选择了化合物45 (代号为Bay 41 2272, IC50 304 nmol·L-1)做了深入研究, 文献中未说明理由, 或许是环丙基具有代谢稳定性。通过机制研究表明45在低水平的一氧化氮下可提高可溶性鸟苷酸环化酶(sGC)的敏感性, 而且当sGC上的卟啉被氧化或除去后, 就失去了刺激产生环鸟苷酸的生成作用。用多种肺动脉高压的动物模型评价了45, 表明有显著降低肺动脉血压, 逆转右心室肥大和肺血管的重塑作用。然而, 由于45显示有抑制并诱导细胞色素P450的作用, 未能进入开发阶段(Straub A, Stasch JP, Alonso-Alija C, et al. NO-independent stimulators of soluble guanylate cyclase. Bioorg Med Chem Lett, 2001, 11: 781-784)。
5 嘧啶环的再优化 5.1 同时评价代谢稳定性和体内活性对新一轮合成的化合物评价体外活性的同时, 还用灌胃清醒的SH大鼠, 测定血压降低10 mmHg的最低有效剂量(MED, mg·kg-1), 作为评价体内活性的指标, 并且测定对CYP 1A2和CYP3A4的作用, 深化优化过程。
5.2 对化合物45和60的深入研究由于尚未探索嘧啶环C6取代的构效关系, 而且4位和6位是对称位置, 因而新一轮重点对45和60合成了4, 6-二氨基化合物, 并与单氨基的做比较, 合成的化合物列于表 5。
| Table 5 Activity and metabolic stability of the compounds with varied substituents at pyrimidine ring. a. MEC: Minimal effective concentration to achieve threefold stimulation of cGMP formation in a recombinant sGC-overexpressing cell line. b. Relaxing effect on pre-contracted rabbit aortic rings. c. MED: Minimal effective dose to induce a mean arterial blood pressure decrease of 10 mm Hg following oral administration to conscious, radiotelemetrically instrumented, spontaneously hypertensive (SH) rats (nd = not determined). d. CYP1A2 inhibition following incubation of phenacetin in the presence of the test compound with human liver microsomes; LC-MS-MS analysis. e. CYP3A4 inhibition following incubation of midazolam in the presence of the test compound with human liver microsomes; pre-incubation was conducted for 30 min; LC-MS-MS analysis of formed metabolites |
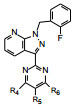

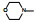
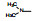
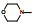
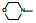
表 5的数据表明环丙基取代的二氨基化合物的体内外活性和对CYP的作用没有明显变化。而吗啉基取代的二氨基化合物66的体内活性提高了10倍, 而且对CYP未显示诱导或抑制作用。然而由于66清除率过快, 且剂量与血药浓度呈非线性关系, 不能进入开发研究。
5.3 C4取代基的变换前已述及, C4的取代基变换对活性影响较小, 因而可以对C4做进一步变换以改善药代动力学性质, 为此同时合成二氨基和单氨基化合物, 表 6列出了化合物的结构与活性。
| Table 6 Activity of the compounds with polar groups at the pyrimidine ring |
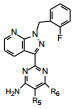
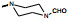
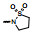
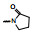
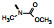
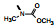

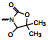
表 6中化合物的C5连接的取代基都有极性基团, 对CYP没有抑制或诱导作用, 故未列出。这些化合物除氨甲酰基72外对sGC酶都有激活作用。化合物71激活sGC酶的作用很强, 但体内降压作用明显弱于66, 故不可取。吡啶取代的两个化合物73和74的体内外活性都较高。含有磺酰基的化合物激活酶的活性虽高, 但对家兔主动脉环和大鼠降压作用平平, 或许是极性过强的缘故。开链的氨基甲酸酯化合物7的活性最强, 对大鼠降压的最低有效剂量为0.1 mg·kg-1, 而其环状类似物81的体内降压作用弱10倍, 增加了偕二甲基取代, 82的体内活性更弱。
5.4 高活性化合物的药代动力学比较从上述高活性的化合物中选择66、73和7做药代动力学性质评价。表 7列出的结果表明化合物7的口服生物利用度显著高于66和73, 血浆半衰期长, 清除率和分布容积低于66和73, 这些都是优良性质。
| Table 7 Pharmacokinetic parameters of three compounds in Beagle dogs |
化合物7具有优良药代动力学性质, 因而深入系统地评价了体内外活性, 在0.1~100 μmol·L-1浓度范围内, 7可浓度依赖性地提高cGMP水平, 最高可达到73倍。与一氧化氮释放剂合用, 显示对激活sGC有协同作用。灌胃清醒大鼠可长时间降低血压。对啮齿类肺动脉高压模型, 可改善血流动力学, 逆转右心室增大和减轻肺动脉的重塑作用。此外, 在10 μmol·L-1浓度下对69种酶没有抑制作用, 表明较少脱靶作用。
|
基于以上优良的药效学和安全性品质, 拜耳公司确定7为候选化合物, 定名为利奥西呱(riociguat), 经3期临床研究, 表明为治疗慢性血栓栓塞性肺动脉高压和肺动脉高压的有效药物, 美国FDA于2013年批准上市(Mittendorf J, Weigand S, Alonso-Alija C, et al. Discovery of riociguat (BAY 63-2521): a potent, oral stimulator of soluble guanylate cyclase for the treatment of pulmonary hypertension. ChemMedChem, 2009, 4: 853-865)。