 2019, Vol. 54
2019, Vol. 54

新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和疗效评价等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等多维性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
介于生物药和化学药之间的肽类药物, 具有高活性和特异性, 是由于与受体发生蛋白-蛋白相互作用, 呈现广泛的互补性结合所致。人体许多激素是内源性多肽, 在执行完生理功能后迅速被代谢失活以避免持续作用而引起不良反应。胰高血糖素样肽-1 (GLP-1)是通过多种机制调控血糖维持血液中葡萄糖稳态的激素, 半衰期很短, 难以直接药用。利拉鲁肽作为GLP-1的改构物, 经系统的氨基酸变换和探索在不同位置连接不同链长的脂链, 提高了活性和稳定性, 成为第一个基于GLP-1结构的降血糖药物。艾塞那肽是蜥蜴唾液中发现的毒蜥外泌肽的合成品, 与GLP-1有相同的生理功能, 虽是GLP-1受体强效激动剂, 但与GLP-1的同源性较低, 因而不被人体水解酶识别, 稳定性强, 成为未做改构直接药用的天然物合成品。肽化学的合成和纯化技术保障了这两个肽类药物的成功上市。关于GLP-1的基础和应用研究是长征性的接力赛, 王明伟等作了全面的综述, 读者可进一步参阅(de Graaf C, Donnelly D, Wootten D, et al. Glucagon-like peptide-1 and its class B G protein–coupled receptors: a long march to therapeutic successes. Pharmacol Rev, 2016, 68: 954-1013)。
(编者按)
胰高血糖素样肽-1 (glucagon-like-peptide-1, GLP-1)是肠内分泌的称作肠促胰素(incretins)的一种肽类激素, 产生于人小肠黏膜的L细胞。GLP-1具有多种生理功能:刺激胰岛的β细胞分泌胰岛素, 抑制高血糖素的释放, 延缓胃的排空, 以及抑制食欲等, 通过降低餐后血糖的漂移, 控制外周血的葡萄糖水平。由于这些效应都是葡萄糖依赖性的, 因而是生理性降低血液中葡萄糖的内源性调节物质。糖尿病患者在消化食物时缺少GLP-1分泌, 导致血糖水平提高。所以, 只要维持一定水平的GLP-1, 理论上应是控制2型糖尿病的有效途径。
1.2 GLP-1的结构与构效关系GLP-1发现于1984年, 是含有30或31个天然氨基酸的肽类激素, 即GLP-1 (7-37)和GLP-1 (7-36)酰胺(Thorens B. Expression cloning of the pancreatic b-cell receptor for the gluco-incretin hormone glucagon-like peptide 1. Proc Natl Acad Sci USA, 1992, 89: 8641-8645)。图 1为GLP-1 (7-37)的结构图。最初认为全长37个氨基酸构成的GLP-1是活性成分, 后来发现N端前6个氨基酸需经剪切后成为7-37的多肽方有活性, 因而沿用了从7开始的编号。

|
Figure 1 The sequence, splicing site, and critical binding sites to receptor (boldface letters) of GLP-1 |
如同人体内所有激素的共同特点一样, 一旦分泌并履行生理功能后, 就被迅速代谢失活, 以维持内环境的稳态。GLP-1也具有快速被降解的性质。在血浆内迅速被二肽基肽酶(DPP-4)水解而失活(因而DPP-4抑制剂可减少GLP-1的失活, 成为口服降糖药物的靶标), 半衰期低于2 min, 酶的剪切位点为Ala8-Glu9, 生成的代谢产物GLP-1 (9-37)和GLP-1 (9-36)酰胺与受体的亲和力很低, 因而失去活性, 甚至有拮抗作用。由于半衰期太短, GLP-1不能直接作为控制糖尿病的药物。
丙氨酸扫描技术研究表明, 组成GLP-1的某些氨基酸是与受体结合并激发功能的重要元件, 例如His7、Gly10、Phe12、Thr13、Asp15、Tyr19、Glu21、Ile29和Val33等关键位点(图 1中用粗体标记), 若被其他氨基酸置换, 会降低或失去活性。这些氨基酸多集中在N端。结构中含有的极性氨基酸残基可作为修饰位点, 以调节GLP-1的物化性质(Knudsen LB. Glucagon-like peptide-1: the basis of a new class of treatment for type 2 diabetes. J Med Chem, 2004, 47: 4128-4134)。
X-射线晶体学研究表明, GLP-1存在两个α螺旋: N端的Thr13-Glu21和C端的Ala24-Val33, C端的螺旋与胞外的受体结合域形成稳定的结合, 而N端螺旋未与胞外区结合, N末端的His7-Phe12的极性片段, 呈无规则的柔性卷曲链, 深入到结合腔内, 是激活受体的主要部分(Runge S, Thøgersen H, Madsen K, et al. Crystal structure of the ligand-bound glucagon-like peptide-1 receptor extracellular domain. J Biol Chem, 2008, 283: 11340-11347)。
1.3 胰高血糖素胰高血糖素样肽-1 (GLP-1)抑制胰高血糖素(glucagon, GCG)的分泌, 二者名词相近, 但功能相反。GCG是体内另一个与糖代谢相关的二十九肽激素, 由胰腺α细胞分泌。胰高血糖素结合于GCG受体, 经一系列生化通路促进糖原分解和降低糖酵解, 使血糖升高, 因而它与胰岛素的作用相反。图 2是胰高血糖素的氨基酸组成, 与GLP-1相比较, 序列上His7、Gly10、Thr11、Phe12、Thr13、Ser14、Asp15、Ser17、Tyr19、Leu20、Ala25、Phe28、Trp31和Leu32是相同的, 同源性为48%, 然而生理功能却完全相反。

|
Figure 2 The structure sequence of glucagon |
评价GLP-1及其衍生物的生物学效果包括两个方面:受体结合的功能性实验和药代动力学的稳定性实验。
2.1 受体功能实验以化合物激动GLP-1受体所生成的c-AMP量作为受试物的活性指标。将克隆表达人GLP-1受体的幼仓鼠肾细胞在DMEM培养液温孵, 匀浆制备细胞膜, 加入不同浓度的受试化合物刺激, 经亲合闪烁方法测定生成的c-AMP, 拟合量效曲线, 计算受试物的活性值EC50。
2.2 药代动力学实验成年猪灌胃受试物0.5 nmol·kg-1 (GLP-1的灌胃剂量为15 nmol·kg-1), 给药前和给药后2、4、6、8、24、48和72 h采集颈静脉血, 用家兔多克隆抗体(抗体对有GLP-1活性的受试物呈特异性结合)放免法测定血样。同时用正常猪血浆混合GLP-1或受试物作为校准实验。温孵3日后加入[125I]GLP-1 (7-36)酰胺, 再温孵。示踪液中加入活性炭悬浮液, 分出结合和游离态抗体。离心, 上清液计数放射性, 经四参数拟合曲线, 计算受试物的浓度。浓度越高受试物的半衰期越长。
Ⅰ 利拉鲁肽 1 缀合长链烃提高与白蛋白的结合力白蛋白是多功能的转运蛋白, 可运载内源性物质和药物。被转运的分子上若连接长链脂肪酸链可提高与白蛋白的结合能力, 从而延长在血浆中的存留时间并降低被肾脏的清除。例如胰岛素的Lys29ξ-氨基被C10~C16脂肪酸酰化, 由于增加了与白蛋白非极性相互作用, 结合力可提高50倍。长脂肪链修饰的胰岛素提高了与白蛋白的结合能力, 延长了胰岛素的降血糖作用, 酰化胰岛素与人、猪和家兔白蛋白的相对结合力为1:1.5:35, 提示人与猪白蛋白的结合力相近, 所以前述的药代评价用猪作模型(Kurtzhals P, Havelund S, Jonassen I, et al. Albumin-binding of insulins acylated with fatty-acids characterization of the ligand protein-interaction and correlation between binding-affinity and timing of the insulin effect in-vivo. Biochem J, 1995, 312: 725-731)。
2 优化目标和结构修饰以GLP-1为起点和参比物, 分别作氨基酸残基变换以及不同位置缀合脂肪酸链, 药效学的目标是, 优化产物应保持或超越GLP-1的活性; 在药动学上, 提高优化产物的代谢稳定性和延长作用时间。优化过程通过衍生物的构效关系和试错实验(trial and error), 内容包括: ①哪(些)个氨基酸可优化出活性更强的多肽; ②在GLP-1肽链的哪个残基可缀合脂肪链而不影响与受体的结合; ③设计怎样的连接基(spacer)保障有效性和稳定性; ④脂肪链长度的优化以确保较长的作用时间。
由于缺乏GLP-1受体的三维结构信息, 因此实施的药物化学方法是依据GLP-1已知各个氨基酸残基的重要性(应用丙氨酸扫描方法确定), 重点在C端引入脂肪链, 避免对参与受体结合和激发活性的N端作过多的修饰。合成的化合物列于表 1中。
| Table 1 Activity of synthetic GLP-1 derivatives |
分析表 1的构效(构代)关系可概括如下: ①在GLP-1分子中仅有两个赖氨酸残基Lys26和Lys34 (不是关键的氨基酸), 所以其他位置的残基都是合成的替换物。赖氨酸侧链有ξ-氨基, 宜于作缀合位点, 所以酰化都发生在Lys残基上。②当C端连接长酰基链, 仍保持高活性, 例如化合物3~9的EC50 30~121 pmol·L-1。化合物2的酰化位点在参与受体结合的8位, 以致活性显著下降到1 260 pmol·L-1。③将GLP-1的两个赖氨酸Lys26和Lys34同时被长链酸酰化(用二元酸或经γ-Glu连接的脂肪链), 如化合物10~13都使活性降低。④在Lys26处缀合不同的长度的脂肪酸链(C12~C18)和不同的连接基(用二元酸或经γ-Glu连接的脂肪链), 得到的化合物活性比较接近, C12、C14的活性一般比C16和C18活性高。⑤化合物19是将His7残基去除氨基与仍保持氨基的His7化合物5活性分别是687 pmol·L-1和61 pmol·L-1, 说明N端的结构变换(尤其是His7)对活性是不利的。⑥在Lys34处连接脂肪酸链的化合物活性一般弱于连接在Lys26的化合物, 如化合物7的活性(121 pmol·L-1)弱于5 (61 pmol·L-1)二倍。化合物23的His7失去了氨基, 活性降低20倍(2 360 pmol·L-1)。⑦将Arg36置换成Lys36并用以连接脂肪酸链, 同时将Lys26, 34变换为Arg26, 34 (赖-精变换), 仍保持活性, 活性的差异取决于脂肪链的大小, 长链的活性弱于短链, 例如化合物27 (C16, 7.89 pmol·L-1)活性强于26 (C20, 210 pmol·L-1), 化合物8 (C16, 36.4 pmol·L-1)的活性强于28 (C18, 116 pmol·L-1), 与前述的规律一致。⑧ GLP-1肽链延长一个Lys38并连接脂肪酸链, 也保持较高活性, 说明远离N端的结构修饰较少影响与受体的结合。但若同时变换关键性氨基酸则会使活性下降, 例如化合物33 (Gly8Arg26, 34)将Ala8变换成Gly8, 活性为328 pmol·L-1, 而相应保持Ala8的化合物9的活性高(53 pmol·L-1)。但变换C端的氨基酸不影响活性, 例如化合物34是将Gly37换成Glu37, 活性(27.2 pmol·L-1)高于化合物9。⑨与Lys相连的脂肪酸链, 以γ-谷氨酸连接基为最优, γ-氨基丁酸也是良好的连接基, 如化合物20与5的活性相近。但β-丙氨酸(化合物21)和吡啶-4-羧酸(22)等连接基的活性减弱。
对表 1中有代表性的高活性化合物测定了药动学(半衰期和生物利用度), 成年猪皮下注射GLP-1和受试物, 表明GLP-1的t1/2 1.2 h, 经长链脂肪酸修饰的化合物t1/2都在9 h以上。表 2列出了化合物的数据。此外, 化合物的生物利用度都在50%以上(数据省略)。
| Table 2 The plasma half life of GLP-1 and high active conjugates |
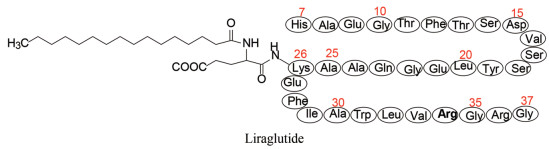
诺和诺德公司基于构效关系分析和综合体外药效学和药代数据, 选择化合物5为候选化合物, 其结构是将Lys34变换成Arg34, 在Lys26的ξ-氨基经谷氨酸连接基, 与十六烷酸连接的GLP-1修饰肽, 定名为利拉鲁肽(liraglutide), 经临床前试验和临床研究, 证明利拉鲁肽具有同GLP-1相同的生理功能, 在血浆中利拉鲁肽因为发生自缔合(self assoxiation)并与白蛋白结合, 显著提高了对DPP-4和内切酶的稳定性, 半衰期t1/2 11~15 h, 患者每日用特制注射器自行皮下注射一次, 可控制2型糖尿病患者的血糖, 成为第一个改构的人GLP-1的降血糖药物, 美国FDA于2010年批准上市。
Ⅱ 艾塞那肽艾塞那肽(37, exenatide)是天然多肽毒蜥外泌肽(exendin-4)的合成品, 由礼来公司研发, 于2005年FDA批准为皮下注射治疗2型糖尿病药物。艾塞那肽作为GLP-1的类似物, 是GLP-1受体完全激动剂, 通过刺激胰岛素分泌, 葡萄糖依赖性地调控血糖, 由于具有代谢稳定性, 是直接作为药用的未经改构的天然多肽合成品。
|
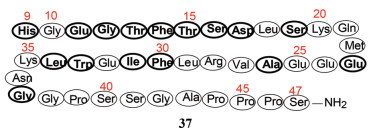
毒蜥外泌肽(exendin-4)是John Eng于1992年在美国退伍军人医学中心研究毒蜥中发现的, 生存于美国西南部的希拉毒蜥(Heloderma suspectum) (图 3)唾液分泌的三十九肽。这种毒蜥的独特习性是食量大, 但进食次数少, 每年仅4次, 食物转化成脂肪, 大量地储存于体内以供不食之需。毒蜥在进食时, 分泌的唾液中既含有毒蜥外泌肽, 也含有GLP-1, 虽然尚不清楚何以同时分泌这两种激素, 但发现都是激活胰岛中的β细胞, 促进胰岛素分泌, 控释葡萄糖和脂肪代谢(Eng J, Kleinman WA, Singh L, et al. Isolation and characterization of exendin-4, an exendin-3 analogue from Heloderma suspectum venom: further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J Biol Chem, 1992, 267: 7402-7405)。毒蜥外泌肽来自爬虫, 与来自哺乳动物的GLP-1在遗传基因上缺乏同源性, 蛋白家族间并无关联, 氨基酸的同源性53% (式37与GLP-1序列相同的氨基酸用粗体标出), 生理功能相同。
|

|
Figure 3 The venom lizard of North America |
毒蜥外泌肽的活性形式是His9-Ser47-NH2, 原肽含有47个氨基酸残基, 前8个需脱除而活化, 故标号自His9开始。毒蜥外泌肽是GLP-1受体的强效激动剂, 结合强度和激动效应强于GLP-1大约5~8倍。作为外源性肽的另一特点是对DPP-4酶有较高的稳定性, 这是毒蜥外泌肽未加修饰直接药用开发成艾塞那肽(以下称艾塞那肽)的原因。
生物物理学和药理学研究表明, 艾塞那肽结构有以下特点: ① N端His9-Asp17氨基酸片段呈无规则的卷曲状态, 是激活受体的主要区域。②分子中形成α螺旋的稳定性强于GLP-1, 是由于在Glu24/Glu25与Arg28之间以及Glu32与Lys35之间形成了分子内氢键, 使螺旋稳固化, 而GLP-1没有相应的分子内氢键。③存在有显著的极性和非极性氨基酸, 这种两性性质增强了与受体胞外域的结合强度。④艾塞那肽比GLP-1多8个氨基酸残基, NMR研究表明, C端的Ser40-Ser48围绕着Trp33形成稳定的笼状构象, 这种构象对受体的结合是有利的。
3 药代动力学性质艾塞那肽与GLP-1的重要区别是N端第二位氨基酸的不同, GLP-1被DPP-4酶特异性水解, 剪切位点是Ala8-Glu9之间的肽键, 艾塞那肽相应的位点是Gly10-Glu11, 后者水解位点的氨基酸改变, 降低了被DPP-4酶的识别和反应, 因而半衰期明显延长。患者皮下注射10 μg 2.1 h后, 达峰浓度Cmax = 211 pg·mL-1, 生物利用度65%~75%, 显著长于GLP-1。艾塞那肽主要经肾小球过滤清除, 清除率为9.1 L·h-1, 半衰期是2.4 h。
4 艾塞那肽的剂量和用法礼来公司研发的艾塞那肽注射液(商品名Byetta)用于改善2型糖尿病患者的血糖控制, 适用于单用二甲双胍、磺酰脲类, 以及二甲双胍合用磺酰脲类, 血糖仍控制不佳的患者。用特制的注射笔皮下注射, 开始剂量为每次5 μg, 每日2次。根据临床应答, 在治疗1个月后剂量可增加至每次10 μg, 每日2次。
Amylin公司研发的长效缓释艾塞那肽剂型(商品名Bydureon)于2012年经FDA批准上市, 为含有2 mg艾塞那肽的微球注射液, 每周注射一次, 控制血糖效果优于每日注射两次的Byetta。