 2019, Vol. 54
2019, Vol. 54



2. 安徽省食品药品检验研究院, 安徽 合肥 230051
2. Anhui Institute for Food and Drug Control, Hefei 230051, China
替硝唑[2-甲基-1-[2-(乙基磺酰基)乙基]-5-硝基-1H-咪唑, tinidazole, 图 1]临床主要用于抗厌氧菌、抗滴虫和阿米巴虫[1]。注射剂型有替硝唑葡萄糖(图 1)注射液, 临床未见严重不良反应报道。

|
Figure 1 Molecular structures of two main drugs |
随着公众与媒体对药物安全性的日益关注, 对药物中杂质的控制已经成为药品质量控制的重要环节。开展杂质谱研究不仅能定性杂质、评价毒性和风险, 而且还能溯源工艺甚至提示工艺改进[2]。
目前, 替硝唑葡萄糖注射液现行标准有《中国药典》2015年版二部和国家食品药品监督管理局药品标准YBH04052012, 在有关物质检查项下均收录了替硝唑的降解杂质2-甲基-5-硝基咪唑, 即杂质1; 另外, 欧洲药典9.0版和美国药典41版还收录了替硝唑的工艺杂质1-(2-乙基磺酰基-乙基)-2-甲基-4-硝基-1H-咪唑, 即杂质5。国内外文献报道较多的是葡萄糖的降解产物5-羟甲基糠醛[3] (杂质2)和杂质1、5[4-6], 其他杂质鲜有报道。
为考察注射剂的一致性评价[7], 本文对替硝唑葡萄糖注射液国评项目中的杂质谱进行比较研究。对93批样品有关物质常规检验后发现, 该品种的杂质谱差别较小, 除部分样品不含杂质6、杂质7外, 其他大多数样品基本包含了替硝唑葡萄糖注射液中的9个杂质(图 2所示编号1~9)。本次实验在310 nm下测得的杂质主要针对大的共轭结构(如芳环、杂环等)或强的增色基团(如卤素碘、磺酰基等), 基本可以包括咪唑环以及替硝唑类似物结构, 但对紫外吸收很弱的葡萄糖骨架难以识别。在210 nm末端吸收的波长下检查葡萄糖的杂质, 由于C18柱对于糖类化合物的分离能力不够而无法达到理想的杂质谱研究。综上, 采用310 nm波长对分离杂质进行定性研究。

|
Figure 2 Impurity profile of tinidazole glucose injection |
通过破坏实验归属杂质来源、推测形成路径, 结合对照品定位、质谱和光谱分析[8-10]、有机反应规律等多种手段, 综合分析并鉴定了替硝唑葡萄糖注射液杂质谱中的2个未知杂质(6和8, 图 2), 它们分别是1-[2-(乙硫氧基)乙基]-2-甲基-5-硝基-1H-咪唑和1, 4-二-N-氧代-2-甲基-5-硝基-1H-咪唑, 为该注射液的杂质毒性研究及安全性评价奠定了基础。
材料与方法仪器 U3000超高效液相色谱仪、Q Exactive Focus四极杆-高分辨轨道阱液质联用仪和Xcalibur 3.0软件(美国Thermo Fisher Scientific公司); LC-20A高效液相色谱仪(二极管阵列检测器, 日本Shimadzu公司); XS105DU电子天平、Seven Multi型pH计(瑞士Mettler Toledo公司)。
试药与试剂 甲醇和乙腈(色谱纯/质谱纯, 美国Thermo Fisher Scientific公司), 纯净水(杭州娃哈哈集团有限公司); 杂质1对照品(2-甲基-5-硝基咪唑, 100512-201603, 中国食品药品检定研究院), 杂质2对照品(5-羟甲基糠醛, 10113667, Alfa Aesar), 杂质5对照品[1-(2-乙基磺酰基-乙基)-2-甲基-4-硝基-1H-咪唑, T1471000, Council of Europe-EDQM CS 30026 F-67081 Strasbourg Cedex], 93批替硝唑葡萄糖注射液(2018年国家药品抽检样品: G217100505, 厂家A; 1711121330, 厂家B; F18010609, 厂家C等), 替硝唑原料(001-171020, 浙江苏泊尔制药有限公司), 葡萄糖原料(P20180208, 潍坊盛泰药业有限公司)。
HPLC色谱条件 采用Thermo C18柱(250 mm×4.6 mm, 5 µm), 以0.05 mol·L-1磷酸二氢钾溶液(磷酸调节pH 3.5)-甲醇(88:12)为流动相, 流速1.0 mL·min-1, 柱温35 ℃, 检测波长310 nm, 进样量20 μL。
UPLC色谱条件 采用Thermo C18柱(100 mm×2.1 mm, 1.9 µm), 含0.1%甲酸的10%甲醇溶液-含0.1%甲酸的乙腈溶液(95:5)为流动相, 流速0.2 mL·min-1, 柱温30 ℃, 检测波长310 nm, 进样量1.0 µL。
质谱条件 离子源为HESI源, 正离子检测模式, 鞘气压力206.8 kPa; 辅助气体积流量10 mL·min-1; 喷雾电压2.00 kV; 离子传输管温度320 ℃; 辅助气温度350 ℃; 扫描模式: Full MS/dd-MS2; Full MS分辨率70 000, dd-MS2分辨率17 500;扫描范围m/z 50~750; MS/MS模式时, 所用碰撞能量为30/40/50 eV。
供试品溶液 精密称取替硝唑0.4 g、葡萄糖5.0 g, 分别置于100 mL量瓶中, 加流动相使溶解并稀释至刻度, 摇匀, 即得替硝唑供试品溶液、葡萄糖供试品溶液。另取替硝唑葡萄糖注射液样品(G217100505, 厂家A)作为替硝唑葡萄糖供试品溶液。
对照品溶液 分别取替硝唑葡萄糖杂质1、2、5对照品适量, 精密称定, 加流动相使溶解并定量稀释成约10 µg·mL-1的溶液作为杂质对照品溶液。
强制降解实验 取上述“供试品溶液” (替硝唑葡萄糖供试品溶液、替硝唑供试品溶液、葡萄糖供试品溶液)各2 mL, 经2 mL 5 mol·L-1稀盐酸溶液60 ℃放置20 h、1 mL 0.1 mol·L-1氢氧化钠室温搅拌30 min、3 mL 30%过氧化氢60 ℃室温放置44 h (2 h、16 h等时间点也取样考察)、105 ℃烘箱放置24 h、光照(4 500 lx)放置4天, 分别处理, 然后加流动相(酸碱处理溶液先中和)稀释至10 mL, 即得强制降解实验溶液。同时进行空白溶剂实验。
结果 1 有关物质检查分析替硝唑葡萄糖供试品、替硝唑供试品、葡萄糖供试品经酸、碱、高温、光照、氧化破坏实验后的色图谱, 结果表明, 替硝唑葡萄糖注射液常见的杂质共有9个(图 2所示编号1~9), 其中杂质1、2、3、4、5、8和9为绝大部分供试品所包含的杂质(杂质4和9的含量水平极低)。注射液中所有批次杂质1含量在0.04%~0.39%之间(均值为0.20%, RSD为0.07%), 其他总杂质含量在0.05%~0.19%之间(均值为0.12%, RSD为0.03%)。替硝唑原料包含杂质1、3、5、6和9, 其中杂质1含量为0.08%, 其他总杂质含量为0.10%。
强制降解实验结果表明, 注射液在碱性和高温下不稳定, 杂质1的含量会明显增加; 在酸性条件下稳定, 仅杂质2 (5-HMF)显著增加; 在氧化条件下主峰峰面积下降, 杂质1和8峰面积增加, 杂质2消失; 光照对产品几乎无影响。
2 杂质结构分析通过UPLC-Q Exactive Focus四极杆-轨道肼高分辨质谱检测得到各杂质的UPLC-UV图(图 3)。使用Xcalibur 3.0软件对高分辨质谱信息进行数据分析, 结合已知杂质对照品定位和质谱解析, 确证各杂质结构, 结果见表 1。

|
Figure 3 UPLC-UV chromatogram of tinidazole and glucose injection |
| Table 1 Impurity structures identified in tinidazole and glucose injection by UPLC-MS/MS |
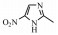


通过杂质对照品定位及质谱定性, 确证杂质1、2、5分别与已知杂质2-甲基-5-硝基咪唑、5-羟甲基糠醛及1-(2-乙基磺酰基-乙基)-2-甲基-4-硝基-1H-咪唑呈一一对应关系。
2.1.1 杂质1测得杂质1一级质谱的准分子离子峰为m/z 128.045 15 [M+H]+, 二级质谱[M+H]+失去1分子NO2形成特征碎片82.053 13 [M-NO2]+, 质谱裂解途径见图 4。根据质谱信息以及杂质对照品定位, 鉴定杂质1为2-甲基-5-硝基咪唑。

|
Figure 4 Fragmentation pathway of impurity 1 [M+H]+ (m/z 128.045 15) ion |
结合强制降解实验结果, 杂质1在碱、氧化、高温破坏条件下含量均显著增加, 特别是在碱破坏实验的优化过程中, 发现随着碱性的增加, 主峰峰面积下降非常明显且几乎完全转化为降解杂质1, 其生成的机制推测:与强吸电子基团(-SO2-, 砜基)相邻的亚甲基在碱性或加热条件下易失去1个H质子, 生成α-碳负离子中间体A, 接着经过负电荷从α-C转移至β-C进而断裂C-N键后, 生成杂质1与乙基乙烯基砜(式中的B); 在310 nm下, 产物B的吸收弱。生成机制见图 5。

|
Figure 5 Proposed formation mechanism of impurity 1 |
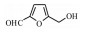
杂质2在一级质谱中出现准分子离子峰m/z 127.038 20 [M+H]+, 在二级质谱中[M+H]+失去1分子H2O形成特征碎片109.028 61 [M-H2O]+, 继续失去1个醛基(CHO)形成特征碎片81.034 02 [M-CHO]+, 质谱裂解途径见图 6。根据质谱信息以及杂质对照品定位及文献报道[2], 鉴定杂质2为5-羟甲基糠醛, 它是单糖的脱水产物, 也是葡萄糖的常见降解杂质。

|
Figure 6 Fragmentation pathways of impurity 2 [M+H]+ (m/z 127.038 20) ion |
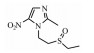
为替硝唑的工艺杂质, 也是替硝唑的同分异构体。杂质5在一级质谱中出现准分子离子峰m/z 248.067 82 [M+H]+, 在二级质谱中[M+H]+砜烷基侧链断裂形成特征碎片128.045 10 [M+H]+和121.032 08 [M+H]+, 质谱裂解途径见图 7。

|
Figure 7 Fragmentation pathway of impurity 5 [M+H]+ (m/z 248.067 82) ion |
根据质谱信息以及杂质对照品定位, 鉴定该杂质为1-(2-乙基磺酰基-乙基)-2-甲基-4-硝基-1H-咪唑。可能的生成机制如图 8所示, 在替硝唑的合成工艺中, 咪唑环上的2个N存在区域选择性的竞争, 导致了羟乙基乙硫醚与2-甲基-5-硝基咪唑取代反应生成两个硫醚异构体, 进而氧化产生了杂质5。

|
Figure 8 Proposed formation mechanism of impurity 5 |
为替硝唑的工艺杂质, 它来源于替硝唑原料合成工艺中的氧化步骤, 性质稳定, 在各破坏实验中含量均未见明显变化。杂质6在一级质谱中出现准分子离子峰m/z 232.073 32 [M+H]+, 在二级质谱中[M+H]+失去1个O之后继而失去一个亚砜侧链产生碎片峰m/z 215.083 62和m/z 110.083 74, 再继而失去1分子NO得到特征碎片m/z 82.053 02, 二级质谱图与可能的裂解途径见图 9。根据正离子模式下一级质谱相对分子质量及二级质谱碎片信息, 推测为1-[2-(乙硫氧基)乙基]-2-甲基-5-硝基-1H-咪唑。

|
Figure 9 MS/MS spectra of impurity 6 [M+H]+ (m/z 232.073 32) ion and its fragmentation pathways |
紫外吸收分光光度法测定最大吸收波长为316 nm, 提示了其与替硝唑具有接近的共轭结构, 此外, 它与替硝唑极性还非常接近(距离替硝唑主峰最近的杂质峰:在UPLC-MS中该杂质保留时间为5.86 min、主峰为6.51 min)。合成工艺(图 10):在钨酸钠/双氧水的氧化下, 硫醚中间体氧化为砜结构的替硝唑过程中, 经历亚砜中间体的过程, 当氧化不完全时, 该亚砜杂质就会生成。亚砜产物与砜产物的极性非常接近, 很多此类反应合成得到的含砜结构类药物, 都涉及亚砜杂质的控制, 如布林佐胺等。

|
Figure 10 Proposed formation mechanism of impurity 6 |
为替硝唑降解为杂质1后继续被氧化的杂质, 存在于几乎所有批次注射液中, 在替硝唑原料及其破坏杂质中未检出。杂质8在一级质谱中出现分子离子峰m/z 159.012 63 [M]+, 根据二级质谱裂解的碎片, 分析该分子离子的裂解经历图示的A、B两条路径(图 11):路径A, 杂质的分子离子失去1个甲基后得到m/z 144.996 64的碎片离子、再失去1个NO2后得到m/z 99.044 50的碎片离子, 该碎片离子具有吡咯环的特征——以逆1, 3-偶极环加成反应的方式失去1个中性分子HCNO后得到m/z 57.034 16的碎片离子; 路径B, 则先失去1分子H2O得到m/z 141.018 07的碎片离子、再失去1个甲基得到m/z 126.986 31的碎片离子、继续失去1个NO2得到m/z 84.960 00的碎片离子, 最后以逆1, 3-偶极环加成反应的方式失去1分子HCN, 同样也得到m/z 57.034 13的碎片离子, 推测为1, 4-二-N-氧代-2-甲基-5-硝基-1H-咪唑。此外, 还观察到该杂质的最大紫外吸收波长为317 nm, 相对于杂质1的308 nm最大吸收波长而言, 发生了明显的红移, 这也能从共轭链得到延长进行解释—杂质8的2个N上以氮-氧p-π反馈键延长了咪唑环的共轭体系, 使得电子云更为离域、能量更低, 所以最大吸收波长数值更大。

|
Figure 11 MS/MS spectra of impurity 8 [M]+ (m/z 159.012 63) ion and its fragmentation pathways |
本研究建立的液质联用方法检测出替硝唑葡萄糖注射液的5个主要杂质, 并通过对各杂质的质谱解析, 综合分析并定性了替硝唑葡萄糖注射液杂质谱中编号的杂质1、2、5、6和8的结构, 其中杂质1、2、5为已经报道过的杂质, 杂质6、8都是未见报道的新化合物。亚砜结构杂质6是合成砜结构替硝唑的“必经之路”, 作为工艺杂质, 它是可以通过改善工艺条件、增加合成工艺中的过程监控及补加氧化剂的方式得以减少甚至避免的。杂质8是注射液中存在(峰面积0.07%), 而替硝唑原料药中不存在的, 并且在氧化破坏条件下杂质8在注射液中含量增加(峰面积0.17%), 而在原料药中依然未检出; 可见, 杂质8更易在溶液状态下产生。需要关注的是, 除了杂质8外, 该注射液和替硝唑原料中的杂质1含量差别也很大, 说明杂质1在溶液状态下更易因主药降解而含量增加。
硝基咪唑结构是具有基因毒性的警示结构片段, 此类硝基咪唑抗菌药(如甲硝唑等)在动物的致癌突变呈阳性, 在人体突变实验呈阴性最终获批上市; 此外, 学术界关于此类药品对人类是否具有致癌突变的争议也一直没有停息过[11]。综上, 值得展望的是, 结构上与替硝唑电子不等排的杂质8, 是否具有更强的基因毒性值得后续研究[12]。
| [1] | Rimbara E, Fischbach LA, Graham DY. Optimal therapy for Helicobacter pylori infections[J]. Nat Rev Gastroenterol Hepatol, 2011, 8: 79–88. DOI:10.1038/nrgastro.2010.210 |
| [2] | Hu CQ. Impurity Profiling of Pharmaceuticals (药物杂质谱分析)[M]. Beijing: Chemical Industry Press, 2015. |
| [3] | Zeng LG. Determination of 5-HMF in tinidazole and glucose injection by HPLC[J]. China Pharm (中国药业), 2010, 19: 18–19. |
| [4] | Naguib IA, Abdelaleem EA, Hassan ES. HPTLC method for simultaneous determination of norfloxacin and tinidazole in presence of tinidazole impurity[J]. J Chromatogr Sci, 2019, 57: 81–86. DOI:10.1093/chromsci/bmy085 |
| [5] | Vaghela BK, Rao SS. A novel validated stability indicating high performance liquid chromatographic method for estimation of degradation behavior of ciprofloxacin and tinidazole in solid oral dosage[J]. J Pharm Bioallied Sci, 2013, 5: 298–308. DOI:10.4103/0975-7406.120082 |
| [6] | Bakshi M. HPLC and LC-MS studies on stress degradation behaviour of tinidazole and development of a validated specific stability-indicating HPLC assay method[J]. J Pharm Biomed Anal, 2004, 34: 11–18. DOI:10.1016/j.japna.2003.08.003 |
| [7] | Tang J, Hu YC. Notice on publicly soliciting opinions on the technical requirements for the conformity evaluation of listed chemical generics (injection)[EB/OL]. 2017-12-22. http://www.cde.org.cn/news.do?method=largeInfo&id=314268. |
| [8] | Wang LU, Wu X, Wang YE, et al. Identification of the related substances of vortioxetine hydrobromide by LC-MS techniques[J]. Acta Pharm Sin (药学学报), 2018, 53: 1351–1356. |
| [9] | Gu X, Luo Y, Chen Y. Identification of fudosteine-related substances by HILIC-MS[J]. Acta Pharm Sin (药学学报), 2017, 52: 1313–1317. |
| [10] | Zhang CY, Li J, Gao JM, et al. The impurity profiling of paclitaxel and its injection by UPLC-MS/MS[J]. Acta Pharm Sin (药学学报), 2016, 51: 965–971. |
| [11] | Bendesky A, Menéndez D. Is metronidazole carcinogenic?[J]. Mutat Res, 2002, 511: 133–144. DOI:10.1016/S1383-5742(02)00007-8 |
| [12] | Gao GH, Yu CR, Li HX, et al. Analysis of control limit of DNA reactive/mutagenic impurities under ICH M7[J]. Chin J New Drugs (中国新药杂志), 2018, 27: 2098–2106. |