 2019, Vol. 54
2019, Vol. 54



2. 宁夏医科大学药学院, 宁夏 银川 750004
2. School of Pharmacy, Ningxia Medical University, Yinchuan 750004, China
转化生长因子-β (transforming growth factor-β, TGF-β)最早于1978年由De Larco与Todaro[1, 2]从鼠肉瘤病毒转化的小鼠成纤维细胞3T3培养液中发现, 因具有刺激正常细胞表型转化活性而命名。迄今在哺乳动物体内已发现有4种不同亚型的TGF-β (TGF-β1~4), 其中TGF-β1活性最强[3]。TGF-β受体有3型, 其中, Ⅰ、Ⅱ型受体属跨膜型丝氨酸/苏氨酸激酶受体, 直接参与细胞内信号转导, Ⅲ型受体不含激酶活性区, 主要起调节TGF-β与Ⅱ型受体结合的作用[4]。TGF-β活化的细胞内信号由Smad和非Smad相关蛋白介导。在TGF-β/Smad信号通路(经典通路)中, TGF-β与Ⅲ型受体(TGFβR Ⅲ)结合, 后者将TGF-β递呈给Ⅱ型受体(TGFβR Ⅱ); 随后, TGF-β与Ⅱ型受体二聚体结合, 进而募集Ⅰ型受体(TGFβR I, 又称ALK5)二聚体形成异源四聚体受体复合物; 此时受体构象发生改变, Ⅱ型受体自身磷酸化, 进而磷酸化并激活Ⅰ型受体; 活化的Ⅰ型受体磷酸化Smad2/Smad3蛋白, 磷酸化的Smad2/Smad3与Smad4结合形成异源复合物并转运至细胞核内[5, 6]。TGF-β介导的非Smad信号通路(非经典通路)则通过上述受体激活丝裂原活化蛋白激酶(MAPK)、磷脂酰肌醇3激酶和蛋白激酶B (PI3K/Akt)、Rho-Rock等通路传导信号入核。其中MAPK通路下游主要包括细胞外信号调节蛋白激酶1/2 (ERK1/2)、c-jun氨基末端激酶(JNK)、p38 MAPK三条信号通路[7]。入核的Smad和/或非Smad信号调节蛋白继而与相应转录因子或辅助蛋白结合, 调控下游靶基因的转录和表达, 从而诱导一系列与多种疾病相关的生物学反应。如通过下调上皮型或血管内皮型钙黏附蛋白(E-cadherin/VE-cadherin)等上皮或内皮标志物表达, 上调纤维连接蛋白(fibronectin)、α平滑肌肌动蛋白(α-SMA)、波形蛋白(vimentin)等间质标志物表达, 使细胞极性消失、运动能力增强, 诱导上皮细胞的上皮-间质转化(epithelial-mesenchymal transition, EMT)或血管内皮细胞的内皮-间质转化(endothelial-mesenchymal transition, EnMT)发生, 从而促进肿瘤侵袭/转移和毛细血管增生。再如, 通过上调fibronectin、α-SMA、胶原蛋白表达, 促成纤维细胞-肌成纤维细胞转分化和细胞外基质(ECM)产生, 促进胶原纤维重塑、机械应力增加, 从而导致组织纤维化。此外, TGF-β还可通过促肿瘤细胞免疫逃逸等加速肿瘤的侵袭和转移[8-10]。鉴于过度活化的TGF-β及其受体所介导的细胞信号通路在肝癌、胰腺癌、骨髓增生异常综合征(myelodysplastic syndromes, MDS)等恶性肿瘤及组织纤维化疾病的发生、发展中发挥关键作用, 一些靶向抑制TGF-β及其受体的小分子抑制剂日益受到关注。其中, 以ALK5为靶的小分子抑制剂品种最为丰富, 这应与ALK5具有高度保守富含甘氨酸-丝氨酸序列的GS结构域(TGFβR Ⅱ为自磷酸激酶, 无GS结构域)和独特的氢键相互作用, 可靶性好, 利于小分子抑制剂的设计、筛选有关[11, 12]。本文结合该领域的关注热点及最新动态, 对TGF-β及其受体小分子抑制剂药物的研究进展作一综述, 为医药工作者提供参考。
1 靶向TGF-β的小分子抑制剂迄今, 明确针对TGF-β配体的小分子抑制剂极少, 吡非尼酮(pirfenidone, PFD)是唯一代表品种。PFD的化学名为5-甲基-1-苯基-2-(1H)-吡啶酮(CAS号: 53179-13-8), 相对分子量(Mr) 185.22, 商品名艾思瑞(Esbriet), 化学结构见图 1。PFD最初由美国Marnac公司开发, 2008年起先后在日本、欧洲、印度上市, 2014年在美国经FDA批准上市, 用于特发性肺纤维化(idiopathic pulmonary fibrosis, IPF)的治疗[13]。PFD可抑制多种细胞中TGF-β的产生, 进而抑制成纤维细胞增殖、减少胶原蛋白合成、延缓IPF病情发展[14]。

|
Figure 1 Structure of pirfenidone |
体外研究结果显示, PFD可抑制貂肺上皮细胞CCL-64中TGF-β前体蛋白转化酶——弗林蛋白酶(furin)的表达, 进而降低TGF-β2 mRNA水平和成熟TGF-β2蛋白的表达[15]。在多种细胞水平模型中, PFD可抑制成纤维细胞的增殖和生物学活性, 对TGF-β诱导的胶原蛋白生成有抑制作用[16, 17]。同时, PFD还能减少人外周血单核细胞产生炎症因子, 如肿瘤坏死因子α (TNF-α)和白细胞介素1β (IL-1β)等[18]。
体内研究结果显示, 在博莱霉素诱导的仓鼠肺纤维化模型中, PFD能降低肺羟脯氨酸表达水平及脯氨酰羟化酶活性, 对肺Ⅰ、Ⅲ型胶原蛋白表达均有抑制作用[19]。大量其他实验动物研究也证明PFD能缓解博莱霉素诱导的肺纤维化症状[20, 21]。在低盐饮食的大鼠模型中, PFD (250 mg·kg-1·d-1, p.o., 治疗28天)可使慢性环孢素诱导的肾纤维化指标改善约50% (P < 0.05), 并使TGF-β1蛋白表达量下降80% (P < 0.001)[22]。另通过多种其他组织纤维化实验动物模型(肺、肝、心脏、肾脏), 证明了PFD具有全身多脏器的抗纤维化活性[23]。
临床试验及上市后治疗观察显示, 对于IPF患者, 与安慰剂组比较, PFD治疗组患者的用力肺活量(FVC)绝对值下降≥10%或病死比例相对降低47.9%, FVC不降低的患者比例相对升高132.5% (P < 0.001)。PFD还可显著缓解6分钟步行距离的减少(P = 0.04)和延长无进展生存期(P < 0.001)。与安慰剂组相比, PFD治疗组在1年内全因死亡的相对风险降低48% (P = 0.01), 1年内IPF相关死亡的相对风险降低68% (P = 0.006)。在超过120周的观察期内, 与安慰剂组相比, PFD治疗组可显著降低IPF患者相关死亡率(P = 0.023 7)[24-26]。
2 靶向TGF-β受体的小分子抑制剂目前, 以TGF-β受体为靶的小分子抑制剂中, ALK5激酶抑制剂研究积累较为深厚, 研制机构众多, 品种丰富, 药用开发方面主要针对肿瘤、MDS、组织纤维化等, 少数品种已进入临床研究阶段。此外, 随着人们对TGF-β受体(TGFβRs)功能认识的加深, 尤其受体蛋白晶体结构的解析, 使部分研究者开始针对配体-受体或受体-受体蛋白之间相互作用设计、筛选新型小分子抑制剂, 从而为寻找针对TGF-β信号通路的小分子抑制剂提供一些新的思路和选择。
2.1 临床研究阶段的ALK5激酶抑制剂现处于临床研究阶段的ALK5激酶抑制剂有3个, 分别是LY2157299、EW-7197和LY3200882 (表 1)。
| Table 1 Small-molecule inhibitors of activin receptor-like kinase 5 (ALK5) in clinical study |
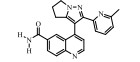

LY2157299 (galunisertib)是美国礼来公司(Eli Lilly and Company)研发的一个ALK5激酶抑制剂, 体外激酶抑制IC50为56 nmol·L-1, 最早于2005年一次国际会议中披露其研究动态[27]。LY2157299作为治疗MDS、脑/肝/胰腺癌、实体瘤的药物开发分别处于Ⅲ期、Ⅱ期和Ⅰ期临床研究阶段, 是目前唯一一个处于临床Ⅲ期试验阶段(NCT02008318)的TGF-β受体小分子抑制剂[28]。
体外研究中, 对受试的人肝癌HLE细胞和HLF细胞, LY2157299可特异性下调TGF-β1诱导的Smad2蛋白磷酸化, 显著抑制癌细胞的增殖和转移。对于初级造血干细胞, LY2157299可剂量依赖性地抑制TGF-β介导的Smad2蛋白活化和造血抑制, 促进原发性MDS患者的骨髓造血功能[29]。体内研究中, 针对TGF-β过度表达的转基因骨髓造血功能衰竭小鼠模型, LY2157299 (100 mg·kg-1·d-1, p.o., 治疗14天)可改善贫血[30]。在人非小细胞肺癌细胞Calu6和乳腺癌细胞MX1异种移植皮下荷瘤裸鼠模型中, LY2157299 (150 mg·kg-1·d-1, p.o., 治疗20天)显示出显著的抗肿瘤活性[31]。
LY2157299治疗MDS的Ⅱ期临床试验结果显示, 疗程内(150 mg, b.i.d., p.o., 治疗14天, 停药14天, 28天一个治疗周期)患者耐受性良好, 单疗程后26%的受试患者达到血液学改善, 输血需求至少降低4个单位或血红蛋白增加至少1.5 g·dL-1, 并维持8周。针对晚期肝癌、神经胶质瘤患者的Ⅰ期、Ⅱ期临床试验结果显示, LY2157299具有良好的药代动力学性质和抗肿瘤疗效, 300 mg·d-1间歇口服给药为安全剂量, 未监测到心血管毒性[32-35]。虽现有观点认为, 长期抑制TGF-β信号转导可能诱发继发性恶性肿瘤, 但LY2157299的临床评价显示, 即使接受治疗超过3年的患者也未观察到肿瘤发生的迹象[36, 37]。
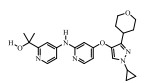
2.1.2 EW-7197EW-7197 (vactosertib)是韩国MedPacto公司研发的ALK5激酶抑制剂, 最早于2012年的一篇会议摘要中报道, 目前正在进行针对MDS的Ⅱ期临床试验(NCT03074006)和晚期实体瘤的Ⅰ期临床试验(NCT02160106)[38, 39]。
体外激酶实验中, EW-7197竞争性结合于ALK5胞内激酶结构域的ATP结合位点产生激酶抑制活性, IC50为11 nmol·L-1。同时, EW-7197还具有ALK4抑制活性, IC50为13 nmol·L-1。在细胞水平上, EW-7197对小鼠乳腺上皮细胞4T1和NMuMG、人乳腺癌上皮细胞MDA-MB-231和MCF10A可剂量依赖性(10~30 nmol·L-1)地阻断TGF-β1诱导的Smad2/Smad3蛋白磷酸化及入核, 进而抑制肿瘤细胞的侵袭和迁移[40]。在4T1、NMuMG、MDA-MB-231肿瘤细胞原位移植小鼠等实验动物模型中, EW-7197 (5~40 mg·kg-1)可抑制TGF-β1诱导的EMT过程。在小鼠乳腺瘤病毒MMTV/c-Neu转基因小鼠模型中, EW-7197 (40 mg·kg-1)可显著抑制乳腺癌的肺转移, 与模型组相比, EW-7197可减少60%的肺转移(P < 0.01)。在4T1肿瘤细胞原位移植小鼠模型中, EW-7197可增强细胞毒性T淋巴细胞(cytotoxic T-lymphocyte, CTL)的活化[41]。在胆管结扎大鼠模型中, EW-7197可通过阻断TGF-β/Smad信号抑制肝纤维化[42]。在小鼠B16黑色素瘤模型中, EW-7197可有效抑制黑色素瘤生长及淋巴结转移。此外, EW-7197除阻断Smad2/Smad3蛋白磷酸化, 还可通过泛素介导的Smad4蛋白降解来增强黑色素瘤小鼠CD8+ T淋巴细胞增生, 进而增强抗黑色素瘤CTL免疫应答[43]。Ⅰ期临床试验结果显示, 对实体瘤患者, EW-7197 (60 mg·d-1, p.o., 治疗28天)尚未发现任何严重的药物相关毒副作用[44, 45]。
2.1.3 LY3200882LY3200882是美国礼来公司研发的另一个高选择性小分子ALK5抑制剂, 可竞争性结合于ALK5激酶结构域的ATP结合位点, 首次报道于2016年ClinicalTrials.gov官网, 目前正在进行针对实体瘤的Ⅰ期临床试验(NCT02937272)[46]。
在体外研究中, LY3200882对受试的肿瘤和免疫细胞均显著抑制TGF-β诱导的Smad蛋白磷酸化。在体外免疫抑制实验中, LY3200882可逆转TGF-β1或调节性T细胞对初始T细胞的抑制活性, 并可恢复后者的增殖能力。体内研究中, 针对三阴乳腺癌4T1-LP原位移植等小鼠模型, LY3200882表现出较强的抗肿瘤生长及转移的活性, 且活性与肿瘤微环境中浸润淋巴细胞的增强有关。此外, 在同系CT26小鼠结肠癌移植瘤模型中, LY3200882与免疫检查点抑制剂PD-L1抗体联合使用, 显示出较好的抗肿瘤活性[47]。
2.2 临床前研究阶段的ALK5激酶抑制剂目前处于临床前研究阶段的ALK5激酶抑制剂品种较多, 包括SB-431542、LY2109761、TP-0427736、IN-1130等十几种小分子抑制剂(表 2)。从已发表的研究数据来看, 研发机构重点关注这些小分子化合物在抑制肿瘤侵袭/转移、控制瘤体毛细血管增生、增强抗瘤免疫应答、控制器官纤维化等方面的药用潜力, 对部分品种与现有抗癌化疗药的体内、外联用活性也较为关注。
| Table 2 Small-molecule inhibitors of ALK5 in preclinical study |
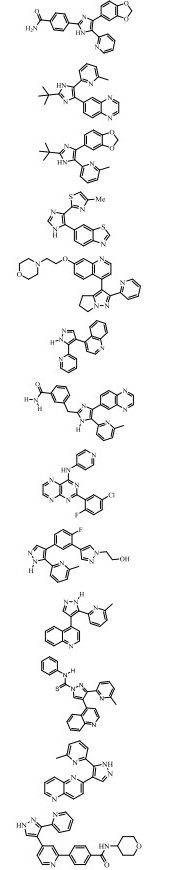
SB-431542是葛兰素史克公司(GlaxoSmithKline)开发的一种强效ALK5抑制剂, 最早报道于2002年。SB-431542体外可抑制ALK5 (IC50 = 94 nmol·L-1)及ALK4、ALK7的激酶活性, 但对ERK、JNK、p38 MAPK信号通路无影响[48, 49]。细胞水平研究显示, 对小鼠成纤维细胞NIH-3T3、胚胎肌母细胞C2C12、乳腺上皮细胞NMuMG以及人永生化表皮细胞HaCaT、肾上皮癌细胞A498、胰腺癌细胞PANC-1等, SB-431542 (≤10 μmol·L-1)可抑制Smad2/Smad3蛋白磷酸化水平、EMT相关生物标志物如fibronectin、α-SMA、Ⅰ型胶原蛋白、Ⅰ型纤溶酶原激活物抑制因子(PAI-1)的转录或表达, 并降低细胞增殖、运动能力[50, 51]。体内研究显示, 在4T1肿瘤细胞移植小鼠乳腺癌模型中, SB-431542 (10 mg·kg-1, i.p., 3次/周, 治疗4周)可显著抑制乳腺癌的肺转移[52]。在小鼠结肠癌模型中, SB-431542 (每只小鼠1×10-4 μmol, i.p., 第3、7天各给药1次)也可激活CTL, 诱导树突状细胞成熟, 产生抗肿瘤免疫应答[53]。该抑制剂在控制肿瘤转移、抑制组织纤维化等方面有潜在应用价值。
2.2.2 LY2109761LY2109761是美国礼来公司研发的一种新型ALK5/TGFβRⅡ双靶点抑制剂, 最早报道于2006年, 体外激酶抑制实验显示其对两个受体的抑制常数Ki分别为38和300 nmol·L-1[54]。在细胞实验中, LY2109761可显著抑制和降低TGF-β1诱导的L3.6pl/GLT细胞迁移、侵袭和细胞中Smad2蛋白磷酸化水平。在体内实验中, LY2109761 (50 mg·kg-1, b.i.d., p.o., 每周一至周五给药), 与吉西他滨(25 mg·kg-1, q.d., i.p., 每周二、五给药)联用4周治疗L3.6pl/GLT荷瘤小鼠; 单用或联用顺铂(50 mg·kg-1, i.p., 1次/周) 4周治疗裸鼠移植瘤(卵巢癌细胞SKOV3或OV-90);单用或联合放疗(每次2 Gy, 第0~4天连续放疗5天; 或每次7 Gy, 第4天单次放疗)治疗(30~40天)两种人成胶质细胞瘤(U87MG和NMA-23)的裸鼠移植瘤, 均显著降低肿瘤体积、延长动物生存期、降低自发性转移、抑制肿瘤微血管形成, 并增强放、化疗效果[55, 56]。目前认为, LY2109761的双激酶抑制活性, 在抗肿瘤活性方面具有独到的优势。
2.2.3 TP-0427736TP-0427736是一种新型ALK5激酶抑制剂, 最早报道于2013年, 体外抑酶IC50为2.72 nmol·L-1, 比抑制ALK3激酶的活性(IC50 = 836 nmol·L-1)高约300倍。体外研究显示, TP-0427736呈浓度依赖性地抑制TGF-β1诱导的A549细胞Smad2/Smad3蛋白磷酸化(IC50 = 8.68 nmol·L-1), 并降低TGF-β对人毛囊外根鞘细胞的生长抑制作用。体内研究显示, TP-0427736能显著抑制小鼠背部皮肤组织Smad2蛋白磷酸化, 也能显著抑制生长后期过渡至退行期引起的毛囊长度缩短, 延长毛囊的生长后期, 从而为雄激素性脱发提供一种潜在的治疗候选物[57, 58]。
2.2.4 IN-1130IN-1130也是一种ALK5抑制剂, 最早报道见于2006年, 其抑制ALK5磷酸化Smad3蛋白的IC50为5.3 nmol·L-1[59]。体外研究显示, 针对人肝癌细胞HepG2, IN-1130可抑制TGF-β诱导的Smad2蛋白磷酸化[60]。体内研究显示, 在4T1肿瘤细胞移植小鼠乳腺癌模型中, IN-1130可抑制TGF-β诱导的磷酸化Smad2蛋白入核、EMT、乳腺癌肺转移[61]。在大鼠单侧输尿管梗阻肾纤维化模型中, IN-1130可抑制肾小管间质性肾炎, 并降低磷酸化Smad2、fibronectin、α-SMA、Ⅰ型胶原蛋白的表达水平, 进而显著抑制肾纤维化过程[59]。
2.2.5 其他ALK5激酶抑制剂除上述4个代表性品种, 表 2中所列其余ALK5激酶抑制剂亦作为药物候选物获得人们相应的关注。如果按照文献报道的体外抑酶IC50值, 将这些分子的激酶抑制活性从高到低做一排序, 可见: R-268712 (2.5 nmol·L-1) > A-83-01 (12 nmol·L-1) > SB-525334 (14.3 nmol·L-1) > GW788388 (18 nmol·L-1) > RepSox (23 nmol·L-1) > A-77-01 (25 nmol·L-1) > SB-505124 (47 nmol·L-1) > SD-208 (48 nmol·L-1) > LY364947 (59 nmol·L-1)。当然, 仅依据体外抑酶活性的强弱, 并不能直接判断这些分子在细胞或整体动物水平抑制TGF-β信号通路的能力。但总体而言, 它们均不同程度对TGF-β诱导的Smad2/Smad3蛋白磷酸化、EMT、肿瘤细胞生长、毛细血管生成、组织纤维化、脱发、疤痕形成等病理过程产生良好的体内外抑制效应, 故吸引大量研发机构进行相关药用研究和评价[62-71]。
2.3 靶向TGF-β-受体相互作用的小分子抑制剂有别于激酶抑制剂的思路, Wang等[72]采用计算机辅助药物设计, 针对TGF-β受体胞外结构域(TGFβR-ECD)三维结构, 基于配体/受体活性口袋的特定位点, 设计、合成了一系列结构新颖的小分子化合物, 代表性品种见表 3。本课题组联合王昊课题组, 从中筛选出一类安全性较高的吲哚类化合物, 通过分子动力学模拟和表面等离子共振(SPR)分析, 发现该类化合物靶向结合于TGF-β1和TGFβRⅡ-ECD。初步研究显示, 这类化合物可有效阻断TGFβRI和TGFβRⅡ的相互作用, 抑制Smad2/Smad3蛋白磷酸化及入核, 并通过相关信号通路在细胞水平抑制上皮细胞EMT过程和成纤维细胞向肌成纤维细胞转分化, 有望在体内抗肿瘤侵袭与转移、抗组织纤维化等方面发挥药理治疗作用。此类新型小分子抑制剂, 以阻断TGF-β及其受体的蛋白-蛋白相互作用(protein-protein interaction, PPI)起始, 进而阻断TGF-β信号转导, 其靶标特异性优于现报道的激酶抑制剂, 具有潜在应用价值。
| Table 3 Small-molecule inhibitors of TGF-β-TGFβR interaction |
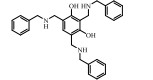

目前, 以TGF-β及受体不同作用位点为靶, 寻找、设计、筛选各种高效、低毒新型小分子抑制剂的研究方兴未艾。如图 2所示, 抑制TGF-β产生的吡非尼酮已成功上市, 而作用于ALK5激酶的LY2157299、EW-7197、LY3200882正处于临床Ⅰ期至Ⅲ期受试者募集或研究评估中。尽管临床前研究阶段的各类候选物分子仍以ALK5激酶抑制剂为主, 但以蛋白-蛋白相互作用位点为靶, 寻找TGF-β信号通路新型抑制剂的思路则颇具启发性。此外, 利用上述小分子抑制剂与其他作用机制药物合理联用, 治疗多种相关疾病也吸引着很多研究者的注意力。总之, 随着新药开发策略的日益丰富, 及安全有效小分子抑制剂的不断开发, 相信会有更多靶向TGF-β及受体的药物进入临床, 发挥应有的治疗作用。

|
Figure 2 Small-molecule inhibitors targeting TGF-β and its receptors |
| [1] | De Larco JE, Todaro GJ. Growth factors from murine sarcoma virus-transformed cells[J]. Proc Natl Acad Sci U S A, 1978, 75: 4001–4005. DOI:10.1073/pnas.75.8.4001 |
| [2] | Roberts AB, Lamb LC, Newton DL, et al. Transforming growth factors: isolation of polypeptides from virally and chemically transformed cells by acid/ethanol extraction[J]. Proc Natl Acad Sci U S A, 1980, 77: 3494–3498. DOI:10.1073/pnas.77.6.3494 |
| [3] | Jenkins G. The role of proteases in transforming growth factor-beta activation[J]. Int J Biochem Cell Biol, 2008, 40: 1068–1078. DOI:10.1016/j.biocel.2007.11.026 |
| [4] | Massagué J. TGF-beta signal transduction[J]. Annu Rev Biochem, 1998, 67: 753–791. DOI:10.1146/annurev.biochem.67.1.753 |
| [5] | Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus[J]. Cell, 2003, 113: 685–700. DOI:10.1016/S0092-8674(03)00432-X |
| [6] | Hinck AP. Structural studies of the TGF-βs and their receptors-insights into evolution of the TGF-β superfamily[J]. FEBS Lett, 2012, 586: 1860–1870. DOI:10.1016/j.febslet.2012.05.028 |
| [7] | Sheen YY, Kim MJ, Park S, et al. Targeting the transforming growth factor-β signaling in cancer therapy[J]. Biomol Ther, 2013, 21: 323–331. DOI:10.4062/biomolther.2013.072 |
| [8] | Chen Q, Yang W, Wang X, et al. TGF-β1 induces EMT in bovine mammary epithelial cells through the TGFβ1/Smad signaling pathway[J]. Cell Physiol Biochem, 2017, 43: 82–93. DOI:10.1159/000480321 |
| [9] | Padua D, Massagué J. Roles of TGF-beta in metastasis[J]. Cell Res, 2009, 19: 89–102. DOI:10.1038/cr.2008.316 |
| [10] | Flavell RA, Sanjabi S, Wrzesinski SH, et al. The polarization of immune cells in the tumour environment by TGF-beta[J]. Nat Rev Immunol, 2010, 10: 554–567. DOI:10.1038/nri2808 |
| [11] | Ge XX, Zhou QF, Chen GL. Advances of transforming growth factor-β inhibitors[J]. Acta Pharm Sin (药学学报), 2015, 50: 413–418. |
| [12] | Ling LE, Lee WC. TGF-beta type Ⅰ receptor (ALK5) kinase inhibitors in oncology[J]. Curr Pharm Biotechnol, 2011, 12: 2190–2202. DOI:10.2174/138920111798808257 |
| [13] | George PM, Wells AU. Pirfenidone for the treatment of idiopathic pulmonary fibrosis[J]. Expert Rev Clin Pharmacol, 2017, 10: 483–491. DOI:10.1080/17512433.2017.1295846 |
| [14] | Maher TM. Pirfenidone in idiopathic pulmonary fibrosis[J]. Drugs Today, 2010, 46: 473–482. DOI:10.1358/dot.2010.46.7.1488336 |
| [15] | Burghardt I, Tritschler F, Opitz CA, et al. Pirfenidone inhibits TGF-beta expression in malignant glioma cells[J]. Biochem Biophys Res Commun, 2007, 354: 542–547. DOI:10.1016/j.bbrc.2007.01.012 |
| [16] | Di SA, Bendia E, Svegliati BG, et al. Effect of pirfenidone on rat hepatic stellate cell proliferation and collagen production[J]. J Hepatol, 2002, 37: 584–591. DOI:10.1016/S0168-8278(02)00245-3 |
| [17] | Lin X, Yu M, Wu K, et al. Effects of pirfenidone on proliferation, migration, and collagen contraction of human Tenon's fibroblasts in vitro[J]. Invest Ophthalmol Visual Sci, 2009, 50: 3763–3770. DOI:10.1167/iovs.08-2815 |
| [18] | Grattendick KJ, Nakashima JM, Feng L, et al. Effects of three anti-TNF-α drugs: etanercept, infliximab and pirfenidone on release of TNF-α in medium and TNF-α associated with the cell in vitro[J]. Int Immunopharmacol, 2008, 8: 679–687. DOI:10.1016/j.intimp.2008.01.013 |
| [19] | Lyer SN, Gurujeyalakshmi G, Giri SN. Effects of pirfenidone on procollagen gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis[J]. J Pharmacol Exp Ther, 1999, 289: 211–218. |
| [20] | Kakugawa T, Mukae H, Hayashi T, et al. Pirfenidone attenuates expression of HSP47 in murine bleomycin-induced pulmonary fibrosis[J]. Eur Respir J, 2004, 24: 57–65. DOI:10.1183/09031936.04.00120803 |
| [21] | Oku H, Shimizu T, Kawabata T, et al. Antifibrotic action of pirfenidone and prednisolone: different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis[J]. Eur J Pharmacol, 2008, 590: 400–408. DOI:10.1016/j.ejphar.2008.06.046 |
| [22] | Shihab FS, Bennett WM, Yi H, et al. Pirfenidone treatment decreases transforming growth factor-β1 and matrix proteins and ameliorates fibrosis in chronic cyclosporine nephrotoxicity[J]. Am J Transplant, 2015, 2: 111–119. |
| [23] | Schaefer CJ, Ruhrmund DW, Pan L, et al. Antifibrotic activities of pirfenidone in animal models[J]. Eur Respir Rev, 2011, 20: 85–97. DOI:10.1183/09059180.00001111 |
| [24] | King TE Jr, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis[J]. N Engl J Med, 2014, 370: 2083–2092. DOI:10.1056/NEJMoa1402582 |
| [25] | Lancaster LH, De JA, Zibrak JD, et al. Pirfenidone safety and adverse event management in idiopathic pulmonary fibrosis[J]. Eur Respir Rev, 2017, 26: 170057. DOI:10.1183/16000617.0057-2017 |
| [26] | Nathan SD, Albera C, Bradford WZ, et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis[J]. Lancet Respir Med, 2017, 5: 33–41. DOI:10.1016/S2213-2600(16)30326-5 |
| [27] | Jonathan Y. Targeting the TGF-β RI kinase with LY2157299: a PK/PD-driven drug discovery and clinical development program[J]. Cancer Res, 2005, 65: 1463. |
| [28] | Eli Lilly and Company. A study of galunisertib in participants with myelodysplastic syndromes[EB/OL]. ClinicalTrials.gov, 2013[2019-03-07]. https://clinicaltrials.gov/ct2/show/NCT02008318. |
| [29] | Dituri F, Mazzocca A, Fernando J, et al. Differential inhibition of the TGF-β signaling pathway in HCC cells using the small molecule inhibitor LY2157299 and the D10 monoclonal antibody against TGF-β receptor type Ⅱ[J]. PLoS One, 2013, 8: e67109. DOI:10.1371/journal.pone.0067109 |
| [30] | Zhou L, Mcmahon C, Bhagat T, et al. Reduced SMAD7 leads to overactivation of TGF-beta signaling in MDS that can be reversed by a specific inhibitor of TGF-beta receptor I kinase[J]. Cancer Res, 2011, 71: 955–963. DOI:10.1158/0008-5472.CAN-10-2933 |
| [31] | Bueno L, Alwis DPD, Pitou C, et al. Semi-mechanistic modelling of the tumour growth inhibitory effects of LY2157299, a new type Ⅰ receptor TGF-β kinase antagonist, in mice[J]. Eur J Cancer, 2008, 44: 142–150. DOI:10.1016/j.ejca.2007.10.008 |
| [32] | Fujiwara Y, Nokihara H, Yamada Y, et al. Phase 1 study of galunisertib, a TGF-beta receptor I kinase inhibitor, in Japanese patients with advanced solid tumors[J]. Cancer Chemother Pharmacol, 2015, 76: 1143–1152. DOI:10.1007/s00280-015-2895-4 |
| [33] | Herbertz S, Sawyer JS, Stauber AJ, et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway[J]. Drug Des Dev Ther, 2015, 9: 4479–4499. |
| [34] | Raymond E, Faivre S, Santoro A, et al. Pharmacokinetics (PK) and pharmacodynamics (PD) of the oral transforming growth factor-beta (TGF-β) receptor I kinase inhibitor LY2157299 monohydrate (LY) in hepatocellular carcinoma (HCC) compared to glioma patients[C]. Mol Cancer Ther, 2013, 12: C261. |
| [35] | Carpentier AF, Brandes AA, Kesari S, et al. Safety interim data from a three-arm phase Ⅱ study evaluating safety and pharmacokinetics of the oral transforming growth factor-beta (TGF-β) receptor I kinase inhibitor LY2157299 monohydrate in patients with glioblastoma at first progression[J]. J Clin Oncol, 2013, 31: 2061. DOI:10.1200/JCO.2012.48.3248 |
| [36] | Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression[J]. Nat Genet, 2001, 29: 117–129. DOI:10.1038/ng1001-117 |
| [37] | Rodon J, Carducci MA, Sepulvedasánchez JM, et al. First-in-human dose study of the novel transforming growth factor-β receptor I kinase inhibitor LY2157299 monohydrate in patients with advanced cancer and glioma[J]. Clin Cancer Res, 2015, 21: 553–560. DOI:10.1158/1078-0432.CCR-14-1380 |
| [38] | MedPacto. Dose escalation and proof-of-concept studies of vactosertib (TEW-7197) monotherapy in patients with MDS[EB/OL]. ClinicalTrials.gov, 2017[2019-03-06]. https://clinicaltrials.gov/ct2/show/NCT03074006. |
| [39] | MedPacto. First in human dose escalation study of vactosertib (TEW-7197) in subjects with advanced stage solid tumors[EB/OL]. ClinicalTrials.gov, 2014[2018-09-05]. https://clinicaltrials.gov/ct2/show/NCT02160106. |
| [40] | Jin CH, Krishnaiah M, Sreenu D, et al. Discovery of N-((4-([1, 2, 4]triazolo[1, 5-a]pyridin-6-yl)-5-(6-methylpyridin-2-yl)-1H-imidazol-2-yl)methyl)-2-fluoroaniline (EW-7197): a highly potent, selective, and orally bioavailable inhibitor of TGF-beta type Ⅰ receptor kinase as cancer immunotherapeutic/antifibrotic agent[J]. J Med Chem, 2014, 57: 4213–4238. DOI:10.1021/jm500115w |
| [41] | Son JY, Park SY, Kim SJ, et al. EW-7197, a novel ALK-5 kinase inhibitor, potently inhibits breast to lung metastasis[J]. Mol Cancer Ther, 2014, 13: 1704–1716. DOI:10.1158/1535-7163.MCT-13-0903 |
| [42] | Kim MJ, Park SA, Kim CH, et al. TGF-β type Ⅰ receptor kinase inhibitor EW-7197 suppresses cholestatic liver fibrosis by inhi-biting HIF1α-induced epithelial mesenchymal transition[J]. Cell Physiol Biochem, 2016, 38: 571–588. DOI:10.1159/000438651 |
| [43] | Yoon JH, Su MJ, Park SH, et al. Activin receptor-like kinase 5 inhibition suppresses mouse melanoma by ubiquitin degradation of Smad4, thereby derepressing eomesodermin in cytotoxic T lymphocytes[J]. EMBO Mol Med, 2013, 5: 1720–1739. DOI:10.1002/emmm.201302524 |
| [44] | Safety Monitoring Committee (SMC) reviewed safety of the first cohort of TEW-7197 anddecided to escalate to the next dose. [EB/OL]. MedPacto News, 2014. http://www.medpacto.com/m/bbs/view.php?idx=9&cate. |
| [45] | Completion of the second cohort of TEW-7197 and SMC decided to escalate to the next dose[EB/OL]. MedPacto News, 2015. http://www.medpacto.com/m/bbs/view.php?idx=10&cate. |
| [46] | Eli Lilly and Company. A study of LY3200882 in participants with solid tumors[EB/OL]. ClinicalTrials.gov, 2016[2019-02-15]. https://clinicaltrials.gov/ct2/show/NCT02937272. |
| [47] | Pei H, Parthasarathy S, Joseph S, et al. LY3200882, a novel, highly selective TGFβRI small molecule inhibitor[J]. Cancer Res, 2017, 77: 955. |
| [48] | Callahan JF, Burgess JL, Fornwald JA, et al. Identification of novel inhibitors of the transforming growth factor beta1 (TGF-beta1) type 1 receptor (ALK5)[J]. J Med Chem, 2002, 45: 999–1001. DOI:10.1021/jm010493y |
| [49] | Inman GJ, Nicolás FJ, Callahan JF, et al. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type Ⅰ activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7[J]. Mol Pharmacol, 2002, 62: 65–74. DOI:10.1124/mol.62.1.65 |
| [50] | Laping NJ, Grygielko E, Mathur A, et al. Inhibition of transforming growth factor (TGF)-beta1-induced extracellular matrix with a novel inhibitor of the TGF-beta type Ⅰ receptor kinase activity: SB-431542[J]. Mol Pharmacol, 2002, 62: 58–64. DOI:10.1124/mol.62.1.58 |
| [51] | Halder SK, Beauchamp RD, Datta PK. A specific inhibitor of TGF-β receptor kinase, SB-431542, as a potent antitumor agent for human cancers[J]. Neoplasia, 2005, 7: 509–521. DOI:10.1593/neo.04640 |
| [52] | Sato M, Matsubara T, Adachi J, et al. Differential proteome analysis identifies TGF-β-related pro-metastatic proteins in a 4T1 murine breast cancer model[J]. PLoS One, 2015, 10: e0126483. DOI:10.1371/journal.pone.0126483 |
| [53] | Tanaka H, Shinto O, Yashiro M, et al. Transforming growth factor β signaling inhibitor, SB-431542, induces maturation of dendritic cells and enhances anti-tumor activity[J]. Oncol Rep, 2010, 24: 1637–1643. |
| [54] | Melisi D, Ishiyama S, Sclabas GM, et al. LY2109761, a novel transforming growth factor beta receptor type Ⅰ and type Ⅱ dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis[J]. Mol Cancer Ther, 2008, 7: 829–840. DOI:10.1158/1535-7163.MCT-07-0337 |
| [55] | Gao Y, Shan N, Zhao C, et al. LY2109761 enhances cisplatin antitumor activity in ovarian cancer cells[J]. Int J Clin Exp Pathol, 2015, 8: 4923–4932. |
| [56] | Zhang M, Kleber S, Röhrich M, et al. Blockade of TGF-β signaling by the TGFβR-I kinase inhibitor LY2109761 enhances radiation response and prolongs survival in glioblastoma[J]. Cancer Res, 2011, 71: 7155–7167. DOI:10.1158/0008-5472.CAN-11-1212 |
| [57] | Amada H, Asanuma H, Koami T, et al. Discovery of 7-methoxy-6-[4-(4-methyl-1, 3-thiazol-2-yl)-1H-imidazol-5-yl]-1, 3-benzothiazole (TASP0382088): a potent and selective transforming growth factor-beta type Ⅰ receptor inhibitor as a topical drug for alopecia[J]. Chem Pharm Bull, 2013, 61: 286–291. DOI:10.1248/cpb.c12-00856 |
| [58] | Naruse T, Aoki M, Fujimoto N, et al. Novel ALK5 inhibitor TP0427736 reduces TGF-β-induced growth inhibition in human outer root sheath cells and elongates anagen phase in mouse hair follicles[J]. Pharmacol Rep, 2017, 69: 485–491. DOI:10.1016/j.pharep.2017.01.024 |
| [59] | Moon JA, Kim HT, Cho IS, et al. IN-1130, a novel transforming growth factor-beta type Ⅰ receptor kinase (ALK5) inhibitor, suppresses renal fibrosis in obstructive nephropathy[J]. Kidney Int, 2006, 70: 1234–1243. DOI:10.1038/sj.ki.5001775 |
| [60] | Lee GT. Effect of IN-1130, a small molecule inhibitor of transforming growth factor-beta type Ⅰ receptor/activin receptor-like kinase-5, on prostate cancer cells[J]. J Urol, 2008, 180: 2660–2667. DOI:10.1016/j.juro.2008.08.008 |
| [61] | Park CY, Min KN, Son JY, et al. A novel inhibitor of TGF-β type Ⅰ receptor, IN-1130, blocks breast cancer lung metastasis through inhibition of epithelial-mesenchymal transition[J]. Cancer Lett, 2014, 351: 72–80. DOI:10.1016/j.canlet.2014.05.006 |
| [62] | Grygielko ET, Martin WM, Tweed C, et al. Inhibition of gene markers of fibrosis with a novel inhibitor of transforming growth factor-beta type Ⅰ receptor kinase in puromycin-induced nephritis[J]. J Pharmacol Exp Ther, 2005, 313: 943–951. DOI:10.1124/jpet.104.082099 |
| [63] | Dacosta BS, Major C, Laping NJ, et al. SB-505124 is a selective inhibitor of transforming growth factor-beta type Ⅰ receptors ALK4, ALK5, and ALK7[J]. Mol Pharmacol, 2004, 65: 744–752. DOI:10.1124/mol.65.3.744 |
| [64] | Li HY, Wang Y, Heap CR, et al. Dihydropyrrolopyrazole transforming growth factor-beta type Ⅰ receptor kinase domain inhibitors: a novel benzimidazole series with selectivity versus transforming growth factor-beta type Ⅱ receptor kinase and mixed lineage kinase-7[J]. J Med Chem, 2006, 49: 2138–2142. DOI:10.1021/jm058209g |
| [65] | Peng SB, Yan L, Xia X, et al. Kinetic characterization of novel pyrazole TGF-beta receptor I kinase inhibitors and their blockade of the epithelial-mesenchymal transition[J]. Biochemistry, 2005, 44: 2293–2304. DOI:10.1021/bi048851x |
| [66] | Ge R, Rajeev V, Ray P, et al. Inhibition of growth and metastasis of mouse mammary carcinoma by selective inhibitor of transforming growth factor-beta type Ⅰ receptor kinase in vivo[J]. Clin Cancer Res, 2006, 12: 4315–4330. DOI:10.1158/1078-0432.CCR-06-0162 |
| [67] | Terashima H, Kato M, Ebisawa M, et al. R-268712, an orally active transforming growth factor-β type Ⅰ receptor inhibitor, prevents glomerular sclerosis in a Thy1 nephritis model[J]. Eur J Pharmacol, 2014, 734: 60–66. DOI:10.1016/j.ejphar.2014.03.045 |
| [68] | Tojo M, Hamashima Y, Hanyu A, et al. The ALK-5 inhibitor A-83-01 inhibits Smad signaling and epithelial-to-mesenchymal transition by transforming growth factor-beta[J]. Cancer Sci, 2010, 96: 791–800. |
| [69] | Gellibert F, Woolven J, Fouchet MH, et al. Identification of 1, 5-naphthyridine derivatives as a novel series of potent and selective TGF-beta type Ⅰ receptor inhibitors[J]. J Med Chem, 2004, 47: 4494–4506. DOI:10.1021/jm0400247 |
| [70] | Gellibert F, de Gouville AC, Woolven J, et al. Discovery of 4-{4--[3-(pyridin-2-yl)-1H-pyrazol-4-yl]pyridin-2-yl}-N-(tetrahydro-2H-pyran-4-yl)benzamide (GW788388): a potent, selective, and orally active transforming growth factor-beta type Ⅰ receptor inhibitor[J]. J Med Chem, 2006, 49: 2210–2221. DOI:10.1021/jm0509905 |
| [71] | Petersen M, Thorikay M, Deckers M, et al. Oral administration of GW788388, an inhibitor of TGF-beta type Ⅰ and Ⅱ receptor kinases, decreases renal fibrosis[J]. Kidney Int, 2008, 73: 705–715. DOI:10.1038/sj.ki.5002717 |
| [72] | Wang H, Richard B, Stephen S, et al. Identification of novel small molecule TGF-beta antagonists using structure-based drug design[J]. J Comput Aided Mol Des, 2013, 27: 365–372. DOI:10.1007/s10822-013-9651-9 |