 2019, Vol. 54
2019, Vol. 54

新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等多维性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
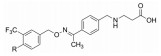
西波莫德是一跟随性创新药物, 诺华公司在自己首创的芬戈莫德上市9年后推出作用于同一靶标和适应症的药物, 是有针对性的再创造:消除药物对S1P3受体脱靶的激动作用, 降低了不良反应; 将羧基药效团组建在结构中, 避免了前药的磷酸化, 因多余的电荷带来不利因素。首创性药物是从无到有, 完全的创新; 跟随性药物需优于或至少不劣于前驱药物, 也包含有创新要素, 本品是一个范例。
(编者按)
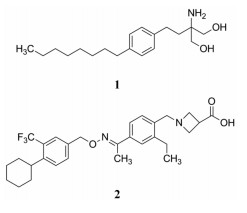
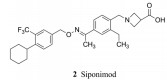
诺华在2010年研发上市的芬戈莫德(1, fingolimod)是作用于鞘氨醇-1-磷酸受体(S1PR)的调节剂, 治疗多发性硬化病, 属于首创性药物。本品西波莫德(2, siponimod)也是诺华研制的, 在2019年上市, 作用于同一靶标, 适应症相同。
|
诺华在9年后推出一款跟随性药物, 显然应超越前驱药物(me-better drug), 公司对后续研究制定了明确的目标, 主要是: ①改变芬戈莫德对S1P的泛激动作用。S1P有5种亚型S1P1~5, 芬戈莫德对5种受体都有活性, 其中对S1P3的激动作用引起心搏徐缓, 该不利的脱靶作用应予以消除; ②芬戈莫德是个前药, 结构中羟基需在体内磷酸化才呈现活性, 磷酸化后对S1PR1呈现拮抗作用为免疫抑制活性, 而且体内活化增加了机体的生化负担和研发成本, 要求酸性基团预构在药物分子中, 并对S1P1呈激动作用; ③在体内活化后的芬戈莫德磷酸酯分子含有两个负电荷, 容易与体内蛋白发生非特异性结合, 导致半衰期过长和分布容积过大, 是不利的药代性质, 换作羧基不仅药代性质可得到改善, 而且也实现了非前药性的结构。
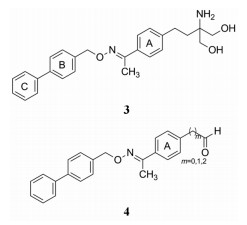
2 先导化合物活性化合物3是诺华对该靶标的另一个苗头化合物, 分子中的极性“头部”与芬戈莫德相同, 但疏水性骨架不同, 为了从苗头3演化出先导化合物(hit-to-lead), 制备了含有胺基和羧基的小型目标库(focused library), 将酸性基团预构在分子之中, 免除体内的活化过程, 以便确定先导物。
合成的策略是制备小型目标库, 用含醛基的中间体4与胺(氨)基酸化合物发生缩合-还原反应而成, 胺基酸的模块见图 1。体外活性评价是用测定受试化合物对放射性S35标记(γ位)的三磷酸鸟苷(GTPγS35)同S1PR1竞争性结合作用来表示(Pan SF, Mi Y, Pally C, et al. A monoselective sphingosine-1-phosphate receptor-1 agonist prevents allograft rejection in a stringent rat heart transplantation model. Chem Biol, 2006, 13: 1227-1234)。

|
Figure 1 Bilding block to condense with 4 for constructing a focused library |
|
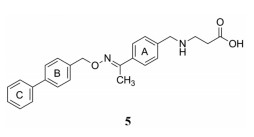
从集中库中筛选出化合物5活性最强, EC50为300 nmol·L-1, 因而5作为先导化合物, 做进一步结构优化。
|
考察联苯基上取代基对S1PR1的活性影响, 合成的化合物和活性列于表 1。构效关系表明: ① B环上3-F (7)或3-CF3 (9)取代活性显著提高, 尤以化合物9为著。而同样基团连接在2位, 如化合物6和8活性显著低于无取代的5, 而且2-CF3的活性非常低。② C环的3'或4'位取代对活性影响不大或降低(10~15); 高活性的化合物9在C环上引入基团(16~19)对活性影响不大, 与9的活性相近。
| Table 1 S1P1 potencies of the compounds with different biphenyl substituents |
上述的构效关系可用分子模拟作诠释。图 2是化合物5对接S1P1的三维投影图。S1P1的结合腔是由带电荷的入口和底部封闭的疏水腔构成。5的羧基负离子与Lys34和Arg120正电荷形成盐键(碱性基团成盐), 还与Tyr29发生氢键作用。5的胺基呈正离子状态, 与Glu121的羧基负离子形成盐键。联苯基进入底部的疏水腔中。从构效关系分析, 环上有3-F取代时可增加疏水相互作用, 因而7的活性比5强。3-CF3还与Leu276和Leu272亲脂性基团发生强力疏水性结合, 因而化合物9的活性甚强于5。然而F或CF3在2位的化合物6和8却因位阻效应而使活性下降。苯环C与B几乎呈垂直取向, 处在疏水腔的底部。

|
Figure 2 Binding mode of compound 5 in S1P1. The electrostatic surface is color coded: blue is positively charged, red is negatively charged, and green is hydrophobic |
基于上述的C环上取代基对活性影响不大的事实, 索性更换C苯环, 例如用杂环或脂肪环替换, 探索对活性的影响, 合成的化合物列于表 2。结果表明, 呋喃(20)和噻吩(21)化合物的活性与9相近, 而环戊基(22)和环己基(23)的活性显著增强, 是9的大约10倍, 而且对S1P3受体的作用很小, 说明脂环的替换物对S1P1受体的选择性较高。因而将环己基作为优化片段固定为分子的疏水末端。
| Table 2 Activity of the compounds with varied terminal rings |
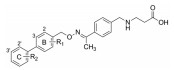
至此, 已经对疏水的两个环片段进行了优化, 下一步主要优化分子另一端的极性基团, 顺便考察苯环B上取代基对活性的影响, 为此合成了化合物2和24~34, 其结构、活性和选择性列于表 3中。
| Table 3 Structures and activity of the compounds with varied polar head groups |
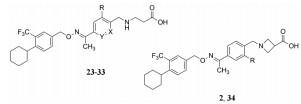

分析构效关系如下: ①苯环A变换为吡啶或噻吩的化合物(24~26)对S1P1受体的活性稍有下降, 并保持对S1P3低活性, 所以没有降低选择性作用。②换成呋喃环(27)的活性显著降低, 推测是呋喃环体积太小(噻吩与苯环相近), 增大了极性头部与疏水尾部之间的两面角所致。③化合物28~33是在末端苯环引入卤素或小尺寸烷基, 对S1P1的活性没有显著影响, 其中31和32是甲基或乙基取代的化合物, 对S1P3的活性更弱, 对提高选择性是有利的基团。
氮杂环丁烷甲酸可视作β-丙氨酸的构象限制物(在α-C和N之间连接CH2), 34和2的活性较强, 尤其是2对S1P1的活性甚强, 选择性也很高, 是最佳优化的化合物(Pan SF, Gray NS, Gao WQ, et al. Discovery of BAF312 (siponimod), a potent and selective S1P receptor modulator. ACS Med Chem Lett, 2013, 4: 333-337)。
4 候选化合物的确定和西波莫德的上市综合高质量的化合物活性、选择性和成药性等性质, 确定了化合物2为初步的候选物, 深入评价了其他生物学性质, 如体外用Caco-2细胞评价2的过膜性质, 表明有良好的被动吸收性, 而且在0.5~50 μmol·L-1浓度范围内未显示有主动外排作用。用多种动物和人的肝微粒体试验代谢作用, 表明有较低或中等程度的代谢转化。体内大鼠和犬的绝对生物利用度分别为50%和71%, 提示胃肠道有较好的吸收性, 而首过效应较低。表 4列出了2的主要药代动力学参数。
| Table 4 In vivo pharmacokinetics in preclinical species |
体内药效学和安全性实验表明2对动物呈现安全有效性, 遂即进入开发阶段, 定名为西波莫德(siponimod), 经Ⅲ期临床研究证明是治疗继发性进展型多发性硬化症的有效药物。于2019年经FDA批准上市(Selmaj K, Li DK, Hartung HP, et al. Siponimod for patients with relapsing-remitting multiple sclerosis (BOLD): an adaptive, dose-ranging, randomised, phase 2 study. Lancet Neurol, 2013, 12: 756-767)。