 2019, Vol. 54
2019, Vol. 54

2. 中国药科大学药物分析教研室, 江苏 南京 210000;
3. 中国药科大学药物化学系, 江苏 南京 210000

 , QU Wei1
, QU Wei1
2. Department of Pharmaceutical Analysis, China Pharmaceutical University, Nanjing 210000, China;
3. Department of Medicinal Chemistry, China Pharmaceutical University, Nanjing 210000, China
癌细胞的增殖需要获得相应的能量和物质基础, 以满足复制的生物合成需求, 同时保持氧化还原稳态。大量研究表明, 肿瘤细胞的代谢异常活跃, 可通过多种途径获取维持其快速增殖所必需的养料及能量[1]。实际上, 癌细胞的代谢也不同于正常细胞[2], 其代谢的改变也被认为是癌症的标志[3]。
癌症代谢不是一个新的领域: 20世纪20年代, Warburg等[4, 5]观察到肿瘤组织较周围的健康组织更快地消耗葡萄糖, 且癌细胞在高速依赖糖酵解的同时能够保证线粒体的呼吸作用。自从这一发现以来, 糖酵解及其他与肿瘤代谢相关的代谢途径得到大量探索[6-8]。近年多项研究表明, 肿瘤细胞中多种氨基酸的代谢水平均发生改变, 以适应能量需求的增加以及肿瘤微环境的变化[9]。
肿瘤细胞氨基酸代谢途径的改变往往由多种信号通路和转录因子共同驱动。大量基础及临床试验研究表明, 靶向肿瘤依赖性氨基酸的代谢, 发展新型药物, 可有效抑制肿瘤的生长[10]。在本综述总结了精氨酸、谷氨酰胺、丝氨酸、色氨酸、天冬酰胺在肿瘤细胞中的代谢途径以及相应的抗肿瘤药物, 简要讨论本领域面临的主要挑战。
1 靶向精氨酸代谢途径与相关的抗肿瘤药物 1.1 肿瘤细胞的精氨酸代谢通路与调控途径精氨酸由瓜氨酸通过精氨琥珀酸合成酶1 (argininosuccinate synthase, ASS1)和精氨琥珀酸裂解酶(argininosuccinate lyase, ASL)的两步催化合成, 然后精氨酸酶1 (arginase, Arg1)将精氨酸分解成鸟氨酸和尿素。通过Arg和鸟氨酸转氨甲酰酶(arginase and ornithine transcarbamylase, OTC)将鸟氨酸转化为瓜氨酸以在线粒体中再循环。其中ASS1、ASL或OTC的异常会影响细胞内精氨酸的储存。在肿瘤中常见ASS1缺乏[11], 导致肿瘤细胞获取血清中的精氨酸。因此, 血清中精氨酸的快速消耗可作为癌症治疗的新策略[12]。精氨酸脱亚胺酶(arginine deiminase, ADI)和Arg1等可以分别通过将精氨酸转化为瓜氨酸和鸟氨酸来耗尽血清中的精氨酸[13, 14], 产生抗肿瘤活性(图 1)。ADI和Arg1介导的抗肿瘤机制包括:诱导细胞周期停滞、细胞凋亡、自噬和抑制血管生成。

|
Figure 1 An overview of the arginine metabolic pathway and plasma arginine depletion for cancer therapy. Clinical trials are currently evaluating ADI-PEG20 and rhArg-PEG for treatment various cancers by depleting plasma arginine |
20世纪60年代, 从关节炎型支原体中获得的ADI首先在体外小鼠淋巴瘤中观察到抗肿瘤活性, 但并未表现出体内活性[15]。随后, 在几种不同类型的人肿瘤中, ADI在体内体外的抗肿瘤作用得到证实[13]。不同菌株的支原体可产生不同类型的ADI。然而在哺乳动物中未发现ADI, 来源于支原体的ADI有很高的抗原性且半衰期短[16]。为解决这个问题, 来源于支原体的ADI与聚乙二醇缀合得到ADI-PEG20, 抗原性显著降低且半衰期明显提高[17]。ADI-PEG20的抗肿瘤活性已在胰腺癌、前列腺癌、小细胞肺癌、淋巴瘤、头颈癌、粘液纤维肉瘤、恶性黑色素瘤、胶质母细胞瘤和乳腺癌中得到证实[18-23]。ADI-PEG20和重组的聚乙二醇化的人精氨酸酶(rhArg1-PEG)通过消耗血清中的精氨酸, 使肿瘤细胞生长受到抑制, 呈现抗肿瘤活性, 抗肿瘤应用与作用机制被广泛研究[12] (表 1)。
| Table 1 Application and regulation mechanism of targeted arginine metabolism drugs in different types of tumors. |
ADI在人体内的免疫原性会影响药物效果[24], 但ADI与其他抗肿瘤药物组合可以改善治疗功效(表 1)。例如, ADI与地塞米松(dexamethasone, DEX)的联合用药对T淋巴细胞白血病细胞以及抗DEX的白血病细胞有协同作用[25]。在前列腺癌细胞中, 多西紫杉醇和ADI-PEG20显示出协同作用[18]。另外, ADI-PEG20可增强培美曲塞在膀胱癌中的活性[26]。最近, ADI-PEG20和多柔比星在乳腺癌中的协同作用被证实[27]。除常规化疗和放疗外, ADI与细胞凋亡诱导剂TRAIL (tumor necrosis factor-related apoptosis-inducing ligand, TRAIL)的联合用药也可协同促进恶性黑色素瘤细胞死亡[28]。因此, 靶向精氨酸代谢药物与其他癌症治疗方式联合在肿瘤的有效治疗上是有前景的。
2 靶向谷氨酰胺代谢途径与相关的抗肿瘤药物 2.1 肿瘤细胞靶向谷氨酰胺代谢与调控途径正常细胞可通过自身合成产生谷氨酰胺, 但肿瘤细胞依靠自身合成的谷氨酰胺不能满足其快速增殖的需要, 需要通过膜上转运体从胞外摄入谷氨酰胺或增强谷氨酰胺代谢通路中关键代谢酶的表达与活性, 以维持细胞增殖的需要。
肿瘤细胞依靠细胞膜上的SLC (solute carrier)超家族转运蛋白从细胞外环境中摄取谷氨酰胺。作为转运谷氨酰胺进入细胞的关键转运体, SLC1A5、SLC7A5、SLC7A11以及SLC6A14在肿瘤细胞中发挥重要作用[35]。已有报道, 转运蛋白SLC6A4在结肠癌、宫颈癌、乳腺癌和胰腺癌中表达上调, 在正常细胞中低水平表达, 阻断SLC6A14蛋白会影响肿瘤细胞内生物合成, 而对正常细胞几乎没影响[36]。Nicklin等[37]发现, SLC1A5的抑制或缺失会导致肿瘤细胞内谷氨酰胺含量降低, mTOR信号通路不能被激活, 最终肿瘤的生长受到抑制。
在线粒体中, 谷氨酰胺经谷氨酰胺酶(glutaminase, GLS)催化成谷氨酸。人体中有两种谷氨酰胺酶的亚型:肾型谷氨酰胺酶(GLS1)和肝型谷氨酰胺酶(GLS2)。在淋巴瘤、神经胶质瘤、乳腺癌、胰腺癌、非小细胞肺癌和肾癌的肿瘤细胞中, 用小分子抑制剂或基因敲除抑制广泛表达的GLS1, 产生抗肿瘤活性[38]。在B淋巴瘤细胞和前列腺癌细胞中, 高表达的致癌转录因子c-Myc可抑制miRNA-23a/b, 上调GLS1的表达, 增强谷氨酰胺代谢, 促进肿瘤细胞的增殖[39]。Wang等[40]在乳腺癌中发现GLS1的上调依赖于Rho家族蛋白和NF-κB信号通路的作用, 并证明GLS1表达的降低能够有效抑制肿瘤细胞的增殖。c-Myc能够诱导GLS1而不诱导GLS2的表达, 而抑癌因子p53诱导GLS2而不诱导GLS1的表达。GLS2在肝细胞癌中表达减少甚至缺失, GLS2过表达可显著减少肿瘤细胞集落的形成, 表明GLS2在抑制肿瘤细胞生长中的潜在作用[41]。
谷氨酰胺经GLS催化产生的谷氨酸, 在线粒体中, 被谷氨酸脱氢酶(glutamate dehydrogenase, GDH)和转氨酶氧化脱氨成α-酮戊二酸和氨, 进入三羧酸循环, 提供能量和物质基础(图 2)。在高度增殖的人乳腺肿瘤细胞中, 转氨酶高表达[42], 且转氨酶表达降低可有效抑制乳腺癌细胞的增殖。GDH可调节肿瘤细胞中α-酮戊二酸的产生, 在肺癌和乳腺癌细胞中表达上调。在人肺癌H1299-异种移植肿瘤裸鼠中, GDH表达的降低可抑制异种移植模型中的肿瘤发生[43]。

|
Figure 2 Glutamine metabolic pathway and target therapy in cancer. Glutamine is imported via SLC1A5, then is hydrolyzed to glutamate by GLS. Glutamate is oxidatively deaminated into α-ketoglutarate(α-KG) through GDH or aminotransferase. α-KG enters the tricarboxylic acid (TCA) cycle. Key enzymes that regulate glutamine metabolism are shown in yellow background in blue, whose target modulators are shown in red |
谷氨酰胺代谢的中间产物参与信号转导。谷氨酸的代谢型谷氨酰胺受体mGlu (metabotropic glutamate receptors)的失活抑制ERK和PI3K信号通路, 促进细胞自噬, 导致肿瘤细胞死亡[44]。抑制谷氨酰胺代谢的中间产物对信号转导通路的调节, 有望治疗癌症。
2.2 靶向谷氨酰胺代谢的药物 2.2.1 抑制关键酶阻断谷氨酰胺代谢途径目前不少针对GLS的抑制剂, 具有抗肿瘤活性。谷氨酰胺类似物, 6-重氮-5-氧代-L-正亮氨酸(6-diazo-5-oxo-L-norlucine, DON, 1, 图 3), 阿西维辛(acivicin, 2, 图 3)通过与酶活性位点的结合成功抑制谷氨酰胺代谢酶, 有抗肿瘤活性, 但因为非选择性和毒性限制其临床应用[45]。双-2-(5-苯基乙酰氨基-1, 2, 4-噻二唑-2-基)乙基硫醚[bis-2-(5-phenylacetamido-1, 2, 4-thiadiazol-2-yl)ethyl sulfide, BPTES, 3, 图 3]为选择性的GLS1变构调节剂, 在体外抑制谷氨酰胺依赖性癌细胞的增殖, 并减缓移植性动物肿瘤和致癌因子c-Myc驱动的小鼠肿瘤的生长[46, 47]。化合物968 (5-(3-溴-4-(二甲基氨基)苯基)-2, 2-二甲基-2, 3, 5, 6-四氢苯并α-菲啶-4 (1H-酮), 4, 图 3)在体外淋巴瘤、乳腺癌、卵巢癌、胶质母细胞瘤和肺癌具有抗肿瘤活性[48-52]。另外, 化合物968可阻断成纤维细胞中各种Rho-GTP酶诱导的致癌转化, 对正常细胞无毒性作用[53]。但由于其疏水性, 很难用于动物模型。目前正在进行临床Ⅰ期试验的CB-839 (5, 图 3), 与BPTES具有相似的变构结合机制和选择性特征, 但表现出更强的抑制活性和独特的动力学性质, 是一种新型的GLS1变构抑制剂, 具有良好的口服生物利用度[38]。另外, CB-839在三阴性乳腺癌(TNBC)细胞系中具有抗增殖活性, 在两种异种移植性肿瘤中显示出显著的抗肿瘤活性[38]。

|
Figure 3 Representative GLS inhibitors |
从FDA (Food and Drug Administration)批准的药物库中鉴定出一种小分子化合物红紫素(purpurin, 6, 图 4), 对纯化的GDH1有显著的抑制作用。但该化合物不能透过细胞发挥抑制作用, 优化后得到可进入细胞的红紫素类似物R162 (7, 图 4), 是有效的靶向GDH1抑制剂, 能够提高癌细胞中的活性氧簇(reactive oxygen species, ROS)水平, 抑制异种移植性肺肿瘤的生长[43]。绿茶茶多酚的主要成分儿茶素(epigallocatechin gallate, EGCG, 8, 图 4)为GDH的抑制剂, 可抑制谷氨酰胺依赖性的乳腺癌细胞的生长[54, 55]。泛转氨酶抑制剂, 氨基氧乙酸(aminooxyacetate, AOA, 9, 图 4), 主要通过激活内质网应激途径抑制高表达c-Myc的乳腺癌细胞系中的谷氨酰化, 导致癌细胞死亡[56]。支链氨基酸转氨酶1 (branched-chain aminotransferase, BCAT1)可调节癌细胞中多种关键蛋白的表达, 抑制BCAT1可降低细胞运动能力且减少细胞外肿瘤微环境的改变, 影响各种肿瘤的转移潜能, 最终发挥抗肿瘤活性[57, 58]。FDA批准的药物加巴喷丁(gabapentin, 10, 图 4)能抑制BCAT1, 其Ki值在毫摩尔范围内。

|
Figure 4 Structures of aminotransferase inhibitors |
谷氨酰胺依靠细胞膜上的转运蛋白进入肿瘤细胞, 抑制相关转运蛋白的表达可阻断谷氨酰胺的摄入, 使肿瘤细胞缺乏能源物质, 最终死亡。
2004年, Esslinger等[59]发现抑制转运蛋白ASCT2 (由SLC1A5基因编码)的一系列化合物, 其中L-γ-谷氨酰-对硝基苯胺(GPNA, 11, 图 5), 在低毫摩尔范围内表现出最好的抑制活性, 但未与ASCT2结合。2012年, Oppedisano等[60]鉴定了第一个结构中不含氨基酸片段的小分子化合物(12, 图 5), 可抑制谷氨酰胺的摄入。Schulte等[61]发现化合物V-9302 [2-氨基-4-双(芳基氧基苄基)氨基丁酸, 13, 图 5]能够强效抑制ASCT2介导的谷氨酰胺转运, 导致癌细胞扩增减少, 氧化损伤增加, 细胞死亡增加, 且在体外和体内均呈现抗肿瘤作用。最新研究发现, 在骨肉瘤细胞和乳腺癌细胞中观察到, V-9302不抑制转运蛋白ASCT2, 而是阻断钠中性氨基酸转运蛋白2 (SNAT2)和大的中性转运蛋白1 (LAT1), 发挥抗肿瘤作用, 并且在非洲爪蟾卵母细胞中重组表达SNAT1、SNAT2、ASCT2和LAT1得到确认[62]。

|
Figure 5 Structures of previous reported inhibitors of ASCT2 and mGlu R1 |
谷氨酰胺代谢过程中产生的谷氨酸, 活化谷氨酸代谢型受体mGlu R1, 从而激活ERK/PI3K/Akt信号通路, 促进肿瘤细胞的增殖[63]。利鲁唑(riluzole, 14, 图 5), 一种治疗肌萎缩性侧索硬化症的药物, 能够抑制mGlu R1, 通过抑制谷氨酸的释放, 阻断信号通路的活化, 抑制三阴性乳腺癌的生长[64]。
3 靶向丝氨酸代谢与相关抗肿瘤药物 3.1 靶向丝氨酰胺代谢与调控途径丝氨酸从头合成途径(de novo serine synthesis pathway, SSP, 图 6)的改变是癌细胞中常见的现象。从糖酵解的中间代谢产物3-磷酸甘油酸(3-phosphoglycerate, 3-PG)开始, 经过涉及3-磷酸甘油酸脱氢酶(3-phosphoglycerate dehydrogenase, PHGDH)、磷酸丝氨酸氨基转移酶(phosphoserine aminotransferase, PSAT) 1和磷酸丝氨酸磷酸酶(phosphoserine phosphatase, PSPH)调控的3个酶促反应产生丝氨酸。SSP中酶表达的升高是癌细胞在丝氨酸饥饿环境能够存活的因素之一。PHGDH在三阴性乳腺癌和黑色素瘤细胞中的表达明显升高, 且抑制PHGDH的表达能够导致肿瘤细胞的增殖率显著下降[65]。PHGDH和PSAT1在非小细胞肺癌中激活, 参与肿瘤的发生发展[66]。另外, 在高转移性乳腺癌细胞系亚型中PSAT1和PSPH高表达[67]。

|
Figure 6 The serine synthesis pathway (SSP) and contribution of serine to one-carbon metabolism. Serine can be synthesized de novo via the SSP in the cytosol. 3-PHP: 3-Phosphohydroxypyruvate; 3-PS: 3-Phosphoserine; THF: Tetrahydrofolate. Reactions involving one-carbon units and GCS system are highlighted with the text '1C' and 'GCS' in blue especially. Key enzymes and regulators are shown in red |
丝氨酸为非必需氨基酸, 抑制丝氨酸的从头合成, 可能引起肿瘤细胞的耐受[68]。寻求外源性丝氨酸代谢中关键酶的抑制剂, 为肿瘤治疗的新方向[69]。外源的丝氨酸通过丝氨酸羟甲基转移酶(serine hydroxymethyltransferases, SHMT1或SHMT2)转化为甘氨酸, 提供一个碳单元参与一碳循环(图 6), 用于核苷酸生物合成。在易患致癌因子Myc驱动的B细胞淋巴瘤的转基因小鼠中观察到SHMT1和SHMT2水平增加[70]。SHMT2是人类肿瘤中最常表达的“代谢基因”之一[71], 敲除SHMT2严重损害癌细胞的增殖。据报道, 甘氨酸裂解系统(glycine cleavage system, GCS)中的甘氨酸脱氢酶(glycine decarboxylase, GLDC)是非小细胞肿瘤中分离的肿瘤起始细胞中最上调的基因之一[72], 抑制GLDC的异常激活可改善非小细胞肺癌患者的存活率。
3.2 靶向丝氨酸代谢药物目前, 很多科研人员开始设计特异性的丝氨酸合成代谢的分子, 实现对肿瘤的抑制。最近, Pacold等[73]和Mullarky等[74]使用高通量筛选鉴定了两种不同的PHGDH小分子抑制剂, 分别是N-(4-甲基吡啶-2-基)-4-(3-(三氟甲基)苯基)哌嗪-1-硫代甲酰胺(PHGDH-hit, 15, 图 7)和5-(呋喃-2-甲酰胺基)-3-甲基-4-硫氰酸噻吩-2-羧酸乙酯(CBR-5884, 18, 图 7)。这两种抑制剂均抑制丝氨酸从头合成, 抑制癌细胞的生长。PHGDH-hit经一系列优化后得到的化合物16、17 (图 7), 化合物17可抑制PHGDH依赖性异种移植的乳腺肿瘤生长, 而不会影响PHGDH非依赖性异种移植瘤[73]。CBR-5884非竞争性和时间依赖性地抑制PHGDH, 并干扰其低聚状态, 可剂量依赖性抑制乳腺癌细胞的生长[74]。

|
Figure 7 Structures of representative inhibitors of PHGDH |
哺乳动物细胞中, L-色氨酸(L-tryptophan, Trp)主要通过“犬尿氨酸途径”分解代谢, 然后转化为L-犬尿氨酸(L-kynurenine, Kyn)。犬尿氨酸途径对Trp的分解代谢受3种不同的双加氧酶控制:色氨酸2, 3-双加氧酶(tryptophan 2, 3-dioxygenase, TDO)、吲哚胺2, 3-双加氧酶1 (indoleamine 2, 3-dioxygenase 1, IDO1)和吲哚胺2, 3-双加氧酶2 (indoleamine 2, 3-dioxygenase 2, IDO2) (图 8)。裂解产物N-甲酰基犬尿氨酸(N-formylkynurenine)经酶促反应或自发水解成犬尿氨酸(L-kynurenine), IDO是催化“犬尿氨酸途径”L-色氨酸分解代谢的限速酶, 并在多种人源肿瘤中高表达[75]。IDO通过消耗肿瘤微环境中对T细胞增殖至关重要的色氨酸, 以及引起色氨酸代谢产物犬尿氨酸及其衍生物的积累, 来抑制先天性和适应性的免疫细胞应答, 导致肿瘤细胞的存活和快速增殖。IDO (IDO1和IDO2)在多种癌症中过表达, 迄今为止专注于IDO1的研究。临床前研究表明, 在啮齿动物模型中靶向IDO1可引发抗肿瘤免疫反应, 抑制肿瘤生长[75]。TDO, 一种同源四聚体血红素B蛋白, 与IDO1、IDO2共同催化犬尿氨酸途径的第一步限速反应, 但TDO对L-色氨酸具有比IDO1更高的亲和力。TDO通常在肝脏中表达, 多在人肿瘤中高表达[76]。临床前研究表明, TDO抑制剂能够促进宿主细胞对肿瘤细胞的免疫排斥, 可作为一种安全有效的癌症治疗方法[77]。

|
Figure 8 The oxidative cleavage of the L-tryptophan by the heme-containing enzymes, IDO1, IDO2, TDO to Nʹ-formylkynurenine, then L-kynurenine. |
1967年, IDO1被首次分离得到, 且其天然底物L-色氨酸在高浓度下能够抑制IDO1的活性, 暗示色氨酸类似物可能具有抑制IDO1的活性[78]。1991年, Cady等[79]报道了两种互为同分异构体的色氨酸类似物: D-1-甲基-色氨酸(D-1-methyl-tryptophan, D-1MT, indoximod, 19, 表 2)和1-甲基-L-色氨酸(L-1-methyl-tryptophan, L-1MT, 20, 表 2), 是IDO1的一种竞争性抑制剂, 能够缓解IDO1对免疫调节激酶mTOR和PKC-Q的抑制作用[80, 81]。D-1MT对IDO1的抑制活性不高, 但与紫杉醇、环磷酰胺等联用时, 可提高这些化学治疗药物的疗效。另外, D-1MT可以抑制啮齿动物肿瘤模型中的肿瘤生长, 同时引发抗癌免疫反应, 已广泛用于研究和临床前研究[75]。
| Table 2 Summary of structures and activity of inhibitors of dioxygenase |


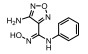
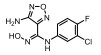


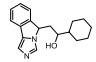
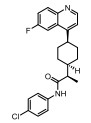
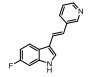
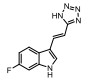
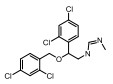
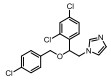
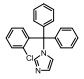
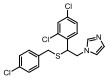
Incyte公司经过高通量筛选获得化合物21 (表 2), 化合物21对IDO1抑制活性的IC50值为1 μmol·L-1, 对TDO抑制浓度的IC50为10 μmol·L-1, 具有明显选择抑制性。同时, 该化合物具有良好的配体效率, 经研究评价后发现可作为良好的先导化合物[82]。该化合物经系列优化后得到羟基脒化合物22、23 (INCB024360, Epacadostat[83], 表 2)。临床前研究表明, 化合物23为IDO1的选择性抑制剂[84], 促进T细胞、NK细胞的生长, 抑制肿瘤细胞。在成功进行Ⅰ期试验后, 目前正在其针对各种恶性肿瘤进行Ⅲ期临床试验。
2011年Zhang等[85]详细介绍IDO1抑制剂Amg-1 (24, 表 2)的结合特性, 从分子模型来看, Amg-1与IDO1的13个残基相互作用, 对IDO1有显著的选择性。2012年, NewLink Genetics公司在一份专利中描述其开发的一系列新的IDO1抑制剂[76]。其中NLG919 (25, 表 2)的抑制活性为75 nmol·L-1, 在临床前模型中引起效应T细胞的剂量依赖性激活和增殖, 导致肿瘤的大规模消退[86, 87]。目前, NLG919在单独干预晚期实体瘤患者中的安全性和初步有效性正在进行临床试验。近日, 百时美施贵宝(Bristol-Myers Squibb, BMS)宣布一种全新的靶向IDO1抑制剂, BMS-986205 (26, 表 2)[88], 具有良好的药代动力学性质, 并能够与其他免疫通路形成互补, 更有效地激活抗肿瘤反应。目前, 与PD-1抗体类药物Nivolumab的联合用药正在进行用于治疗黑色素瘤的临床试验。1995年, Salter等[89]描述了一种新型氟吲哚680C91 (27, 人和鼠TDO的IC50分别为880和55 nmol·L-1, 表 2), 为TDO和5-羟色胺摄取抑制剂, 与IDO1不发生交叉反应, 但在体内表现出较差的水溶性和口服生物利用度。为寻找具有良好药物性质的特异性TDO抑制剂, 2011年Dolušić团队[90]对TDO抑制剂进行合理的药物设计, 经过对70多种化合物进行评估, 确定(E)-6-氟-3-[2-(1H-四唑-5-基)乙烯基]-1H-吲哚(LM10, 28, 人TDO: IC50 = 5.6 nmol·L-1, 表 2)为水溶性, 得到改善的选择性TDO抑制剂(IDO1: IC50 > 400 μmol·L-1)。目前, TDO抑制剂在临床前研究中表现出较好的抗肿瘤活性[91]。Pilotte等[77]表明, 抑制TDO和IDO1以提高癌症免疫疗法的效率将是互补的, 而不是多余的。研究结果也表明, TDO抑制剂可通过促进免疫排斥, 从而利于免疫癌症的治疗[77]。
IDO2为IDO1的旁系同源物, 他们具有相似的基因组结构和血红素环境[92]。然而, IDO1和IDO2呈现出不同的底物特异性和组织分布。IDO2在多种癌症中的表达已经得到证实[93]。另外, IDO2是诱导几种关键炎性细胞因子所必需的, 且靶向IDO1和IDO2能够增强免疫效应[94]。在寻找IDO2抑制剂时, Bakmiwewa等[91]发现一系列抗真菌咪唑类化合物(29~32, 表 2)可有效地抑制IDO1、IDO2和TDO, 且对细胞活力影响不大。因此, 在抑制所有色氨酸-犬尿氨酸代谢酶的方面, 抗真菌咪唑类化合物能够与芳烃受体AhR (aryl hydrocarbon receptor)和细胞色素P450超家族相互作用, 相比于其他发表的化合物表现出更有效的抑制活性, 代表未来研究的新途径[95, 96]。
5 天冬酰胺代谢途径与抗肿瘤药物哺乳动物细胞通过天冬酰胺合成酶(asparagine synthetase, ASNS)从天冬氨酸和谷氨酰胺产生天冬酰胺。然而, 一些癌细胞缺乏ASNS的表达, 需要依赖血清中的天冬酰胺满足需要。
目前, 第一个直接靶向天冬氨酸代谢的抗肿瘤药物是来自大肠杆菌和欧洲野花欧文氏菌的L-天冬酰胺酶(L-asparaginase, L-ASPase), 经FDA批准用于治疗儿科和成人急性淋巴细胞白血病(acute lymphoblastic leukemia, ALL)。L-ASPase广泛存在于微生物、植物和部分锯齿类动物血清中, 经微生物发酵法生产得到的酶抑制剂。免疫原性降低并半衰期延长的聚乙二醇化大肠杆菌L-ASPase也被FDA批准用于ALL治疗。目前许多临床试验正在评估L-ASPase治疗一系列恶性血液肿瘤的疗效。L-ASPase催化天冬酰胺的脱酰胺作用[97], 快速消耗血清中天冬酰胺, 导致肿瘤细胞蛋白质合成受阻, 生长受到抑制。正在临床中使用的大肠杆菌(Escherichia coli)和欧文菌(Erwinia chrysanthemi) Ⅱ型L-ASPase在体内不稳定, 半衰期短, 需要几次给药以保证活性浓度。此外, 它们对耐药的白血病淋巴母细胞过表达的组织蛋白酶B和天冬酰胺内肽酶敏感, 从而影响药物活性和药代动力学性质。研究显示, 新的欧文氏菌Ⅱ型ASN酶变体N24S, 一种蛋白酶抗性大肠杆菌天冬酰胺酶, 具有突出的稳定性和增强的体外抗白血病活性, 成为一种潜在的替代疗法[98]。
6 靶向氨基酸代谢药物开发面临的挑战随着肿瘤氨基酸代谢基础研究的不断深入以及靶点发现手段的成熟, 针对氨基酸代谢的小分子抑制剂的发现也日益增多。然而成功上市的靶向氨基酸代谢的抗癌药较少。其原因可能是多方面的, 主要体现在以下几个方面:
6.1 氨基酸代谢影响抗肿瘤免疫应答免疫细胞在静止状态与激活状态下对于能量的利用具有明显差异, 代谢激活时, 对营养物质的吸收增加, 最终正常发挥免疫应答。然而, 实验发现, 当肿瘤细胞内精氨酸代谢关键酶不足时, 肿瘤微环境中的肿瘤相关的骨髓细胞可提供肿瘤细胞对精氨酸的需求[99, 100]。同时, T细胞也需要消耗大量的微环境中的精氨酸来发挥功能, 增加抗肿瘤免疫应答[101]。因此, 靶向抑制血清中的精氨酸代谢在杀伤癌细胞的同时也会抑制免疫细胞的功能。另外, 肿瘤细胞大量消耗微环境中的色氨酸, 导致效应T细胞凋亡[102, 103]。因此, 靶向肿瘤细胞氨基酸代谢会影响抗肿瘤的免疫细胞功能, 在抗肿瘤和维持免疫应答之间找到合适靶向的氨基酸代谢药物, 需要明确肿瘤细胞特有的代谢通路。另外, 癌细胞会在缺乏营养素吸收等信号时激活大分子的自噬降解, 降解后产生的小分子营养物质以供给代谢所需。
6.2 氨基酸代谢的表型差异大当细胞在体内和离体模型系统之间转换时, 代谢表型会发生改变。突变KRAS驱动的非小细胞肺癌对外源谷氨酰胺的利用很少, 但来自这些肿瘤的培养细胞系则依赖于外部谷氨酰胺供应, 对谷氨酰胺酶(GLS)的抑制也很敏感。
另外, 肿瘤类型不同, 其氨基酸代谢的表型存在差异。Myc诱导的肺肿瘤中GLUL上调, 然而在肝肿瘤中GLUL表达降低。甚至不同的癌症亚型也显示出不同的代谢表型。例如, 基底乳腺癌细胞通常依赖谷氨酰胺并且对GLS抑制敏感, 而大多数管腔乳腺癌细胞表达GLUL, 对GLS抑制剂具有抗性[38, 104]。
6.3 特定模型系统的局限性氨基酸在健康组织和肿瘤患者中的浓度有明显差异, 然而目前氨基酸代谢领域的研究大多依赖于在标准培养基中生长的癌细胞系。血清中谷氨酰胺的浓度在健康个体中约为0.5 mmol·L-1, 在许多癌症患者中较低, 而标准细胞培养基含有2~4 mmol·L-1谷氨酰胺[105]。另外, 大多数氨基酸在脑肿瘤中的浓度比血清低2~12倍[106]。
因氨基酸代谢固有的灵活性、冗杂性以及当前肿瘤类型多而模型系统的局限性, 为靶向氨基酸代谢药物的发现提出了挑战。因此, 研究时选择适合的肿瘤类型、培养方式和模型系统, 对于靶向氨基酸代谢药物的成功开发至关重要。
7 结语靶向氨基酸代谢的药物作为个性化癌症治疗的组分, 已经从临床前研究发展到临床试验, 并且在某些情况下显示出功效。本综述对干预肿瘤细胞氨基酸代谢的几种靶向药物进行总结, 希望随着对肿瘤氨基酸代谢更广泛理解以及分析代谢技术的进步, 能够精准地选择合适的模型系统和识别目标患者, 以期为靶向氨基酸代谢的新型癌症治疗药物的发现提供思路和参考。
| [1] | Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism[J]. Cell Metab, 2016, 23: 27–47. DOI:10.1016/j.cmet.2015.12.006 |
| [2] | Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism[J]. Nat Rev Cancer, 2011, 11: 85–95. DOI:10.1038/nrc2981 |
| [3] | Cantor JR, Sabatini DM. Cancer cell metabolism: one hallmark, many faces[J]. Cancer Discov, 2012, 2: 881–898. DOI:10.1158/2159-8290.CD-12-0345 |
| [4] | Warburg O, Wind F, Negelein E. The metabolism of tumors in the body[J]. J Gen Physiol, 1927, 8: 519–530. DOI:10.1085/jgp.8.6.519 |
| [5] | Warburg O. On the origin of cancer cells[J]. Science, 1956, 123: 309–314. DOI:10.1126/science.123.3191.309 |
| [6] | Choi J, Kim ES, Koo JS. Expression of pentose phosphate pathway-related proteins in breast cancer[J]. Dis Markers, 2018, 2018: 9369358. |
| [7] | Shuvalov O, Petukhov A, Daks A, et al. One-carbon metabolism and nucleotide biosynthesis as attractive targets for anticancer therapy[J]. Oncotarget, 2017, 8: 23955–23977. |
| [8] | Santos CR, Schulze A. Lipid metabolism in cancer[J]. FEBS J, 2012, 279: 2610–2623. DOI:10.1111/ejb.2012.279.issue-15 |
| [9] | Hanahan D, Weinberg Robert A. Hallmarks of cancer: the next generation[J]. Cell, 2011, 144: 646–674. DOI:10.1016/j.cell.2011.02.013 |
| [10] | Lukey MJ, Katt WP, Cerione RA. Targeting amino acid metabolism for cancer therapy[J]. Drug Discov Today, 2016, 6: 281–289. |
| [11] | Dillon BJ, Prieto VG, Curley SA, et al. Incidence and distribution of argininosuccinate synthetase deficiency in human cancers[J]. Cancer, 2004, 100: 826–833. DOI:10.1002/(ISSN)1097-0142 |
| [12] | Qiu F, Huang J, Sui M. Targeting arginine metabolism pathway to treat arginine-dependent cancers[J]. Cancer Lett, 2015, 364: 1–7. DOI:10.1016/j.canlet.2015.04.020 |
| [13] | Takaku H, Takase M, Abe SI, et al. In vivo anti-tumor activity of arginine deiminase purified from Mycoplasma arginine[J]. Int J Cancer, 1992, 51: 244–249. DOI:10.1002/(ISSN)1097-0215 |
| [14] | Sezgin N, Torun T, Yalcin F. Biochemical characterization of the arginine degrading enzymes arginase and arginine deiminase and their effect on nitric oxide production[J]. Med Sci Monit, 2002, 8: BR248–BR253. |
| [15] | Kraemer PM, Defend V, Hayflick L, et al. Mycoplasma (PPLO) strains with lytic activity for murine lymphoma cells in vitro[J]. Proc Soc Exp Biol Med, 1963, 112: 381–387. DOI:10.3181/00379727-112-28052 |
| [16] | Ni Y, Schwaneberg U, Sun ZH. Arginine deiminase, a potential anti-tumor drug[J]. Cancer Lett, 2008, 261: 1–11. DOI:10.1016/j.canlet.2007.11.038 |
| [17] | Holtsberg FW, Ensor CM, Steiner MR, et al. Poly (ethylene glycol) (PEG) conjugated arginine deiminase: effects of PEG formulations on its pharmacological properties[J]. J Control Release, 2002, 80: 259–271. DOI:10.1016/S0168-3659(02)00042-1 |
| [18] | Kim RH, Coates JM, Bowles TL, et al. Arginine deiminase as a novel therapy for prostate cancer induces autophagy and caspase-independent apoptosis[J]. Cancer Res, 2009, 69: 700–708. DOI:10.1158/0008-5472.CAN-08-3157 |
| [19] | Syed N, Langer J, Janczar K, et al. Epigenetic status of argininosuccinate synthetase and argininosuccinate lyase modulates autophagy and cell death in glioblastoma[J]. Cell Death Dis, 2013, 4: e458. DOI:10.1038/cddis.2012.197 |
| [20] | Huang HY, Wu WR, Wang YH, et al. ASS1 as a novel tumor suppressor gene in myxofibrosarcomas: aberrant loss via epigenetic DNA methylation confers aggressive phenotypes, negative prognostic impact, and therapeutic relevance[J]. Clin Cancer Res, 2013, 19: 2861–2872. DOI:10.1158/1078-0432.CCR-12-2641 |
| [21] | Huang CC, Tsai ST, Kuo CC, et al. Arginine deprivation as a new treatment strategy for head and neck cancer[J]. Oral Oncology, 2012, 48: 1227–1235. DOI:10.1016/j.oraloncology.2012.06.004 |
| [22] | Feun LG, Marini A, Walker G, et al. Negative argininosuccinate synthetase expression in melanoma tumors may predict clinical benefit from arginine-depleting therapy with pegylated arginine deiminase[J]. Br J Cancer, 2012, 106: 1481–1485. DOI:10.1038/bjc.2012.106 |
| [23] | Bowles TL, Kim R, Galante J, et al. Pancreatic cancer cell lines deficient in argininosuccinate synthetase are sensitive to arginine deprivation by arginine deiminase[J]. Int J Cancer, 2008, 123: 1950–1955. DOI:10.1002/ijc.v123:8 |
| [24] | Glazer ES, Piccirillo M, Albino V, et al. Phase Ⅱ study of pegylated arginine deiminase for nonresectable and metastatic hepatocellular carcinoma[J]. J Clin Oncol, 2010, 28: 2220–2226. DOI:10.1200/JCO.2009.26.7765 |
| [25] | Noh EJ, Kang SW, Shin YJ, et al. Arginine deiminase enhances dexamethasone-induced cytotoxicity in human T-lymphoblastic leukemia CCRF-CEM cells[J]. Int J Cancer, 2004, 112: 502–508. DOI:10.1002/(ISSN)1097-0215 |
| [26] | Allen MD, Luong P, Hudson C, et al. Prognostic and therapeutic impact of argininosuccinate synthetase 1 control in bladder cancer as monitored longitudinally by PET imaging[J]. Cancer Res, 2014, 74: 896–907. DOI:10.1158/0008-5472.CAN-13-1702 |
| [27] | Qiu F, Chen YR, Liu X, et al. Arginine starvation impairs mitochondrial respiratory function in ASS1-deficient breast cancer cells[J]. Sci Signal, 2014, 7: ra31. DOI:10.1126/scisignal.2004761 |
| [28] | You M, Savaraj N, Wangpaichitr M, et al. The combination of ADI-PEG20 and TRAIL effectively increases cell death in melanoma cell lines[J]. Biochem Biophys Res Commun, 2010, 394: 760–766. DOI:10.1016/j.bbrc.2010.03.066 |
| [29] | Kim JE, Kim SY, Lee KW, et al. Arginine deiminase originating from Lactococcus lactis ssp. lactis American Type Culture Collection (ATCC) 7962 induces G1-phase cell-cycle arrest and apoptosis in SNU-1 stomach adenocarcinoma cells[J]. Br J Nutr, 2009, 102: 1469–1476. DOI:10.1017/S0007114509990432 |
| [30] | Haines RJ, Pendleton LC, Eichler DC. Argininosuccinate synthase: at the center of arginine metabolism[J]. Int J Biochem Cell Biol, 2011, 2: 8–23. |
| [31] | Delage B, Luong P, Maharaj L, et al. Promoter methylation of argininosuccinate synthetase-1 sensitises lymphomas to arginine deiminase treatment, autophagy and caspase-dependent apoptosis[J]. Cell Death Dis, 2012, 3: e342. DOI:10.1038/cddis.2012.83 |
| [32] | Sicinschi LA, Lopezcarrillo L, Camargo MC, et al. Gastric cancer risk in a Mexican population: role of Helicobacter pylori CagA positive infection and polymorphisms in interleukin-1 and -10 genes[J]. Int J Cancer, 2010, 118: 649–657. |
| [33] | Lam TL, Wong GKY, Chong HC, et al. Recombinant human arginase inhibits proliferation of human hepatocellular carcinoma by inducing cell cycle arrest[J]. Cancer Lett, 2009, 277: 91–100. DOI:10.1016/j.canlet.2008.11.031 |
| [34] | Morrow K, Hernandez CP, Raber P, et al. Anti-leukemic mechanisms of pegylated arginase I in acute lymphoblastic T-cell leukemia[J]. Leukemia, 2013, 27: 569–577. DOI:10.1038/leu.2012.247 |
| [35] | Bhutia YD, Babu E, Ramachandran S, et al. Amino acid transporters in cancer and their relevance to "glutamine addiction": novel targets for the design of a new class of anticancer drugs[J]. Cancer Res, 2015, 75: 1782–1788. DOI:10.1158/0008-5472.CAN-14-3745 |
| [36] | Coothankandaswamy V, Cao S, Xu Y, et al. Amino acid transporter SLC6A14 is a novel and effective drug target for pancreatic cancer[J]. Br J Pharmacol, 2016, 173: 3292–3306. DOI:10.1111/bph.13616 |
| [37] | Nicklin P, Bergman P, Zhang B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy[J]. Cell, 2009, 136: 521–534. DOI:10.1016/j.cell.2008.11.044 |
| [38] | Gross MI, Demo SD, Dennison JB, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer[J]. Mol Cancer Ther, 2014, 13: 890–901. DOI:10.1158/1535-7163.MCT-13-0870 |
| [39] | Gao P, Tchernyshyov I, Chang TC, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism[J]. Nature, 2009, 458: 762–765. DOI:10.1038/nature07823 |
| [40] | Wang JB, Erickson JW, Fuji R, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation[J]. Cancer Cell, 2010, 18: 207–219. DOI:10.1016/j.ccr.2010.08.009 |
| [41] | Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy[J]. Nat Rev Cancer, 2016, 16: 619–634. DOI:10.1038/nrc.2016.71 |
| [42] | Coloff JL, Murphy JP, Braun CR, et al. Differential glutamate metabolism in proliferating and quiescent mammary epithelial cells[J]. Cell Metab, 2016, 23: 867–880. DOI:10.1016/j.cmet.2016.03.016 |
| [43] | Jin L, Li D, Alesi N, et al. Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth[J]. Cancer Cell, 2015, 27: 257–270. DOI:10.1016/j.ccell.2014.12.006 |
| [44] | Zhang C, Yuan XR, Li HY, et al. Anti-cancer effect of metabotropic glutamate receptor 1 inhibition in human glioma U87 cells: involvement of PI3K/Akt/mTOR pathway[J]. Cell Physiol Biochem, 2015, 35: 419–432. DOI:10.1159/000369707 |
| [45] | Lukey MJ, Wilson KF, Cerione RA. Therapeutic strategies impacting cancer cell glutamine metabolism[J]. Future Med Chem, 2013, 5: 1685–1700. DOI:10.4155/fmc.13.130 |
| [46] | Wang JB, Erickson JW, Fuji R, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation[J]. Cancer Cell, 2010, 18: 207–219. DOI:10.1016/j.ccr.2010.08.009 |
| [47] | Xiang Y, Stine ZE, Xia J, et al. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis[J]. J Clin Invest, 2015, 125: 2293–2306. DOI:10.1172/JCI75836 |
| [48] | Simpson NE, Tryndyak VP, Beland FA, et al. An in vitro investigation of metabolically sensitive biomarkers in breast cancer progression[J]. Breast Cancer Res Treat, 2012, 133: 959–968. DOI:10.1007/s10549-011-1871-x |
| [49] | Martínrufián M, Nascimentogomes R, Higuero A, et al. Both GLS silencing and GLS2 overexpression synergize with oxidative stress against proliferation of glioma cells[J]. J Mol Med, 2014, 92: 277–290. DOI:10.1007/s00109-013-1105-2 |
| [50] | Mohamed A, Deng X, Khuri FR, et al. Altered glutamine metabolism and therapeutic opportunities for lung cancer[J]. Clin Lung Cancer, 2014, 15: 7–15. DOI:10.1016/j.cllc.2013.09.001 |
| [51] | Simpson NE, Tryndyak VP, Pogribna M, et al. Modifying metabolically sensitive histone marks by inhibiting glutamine metabolism affects gene expression and alters cancer cell phenotype[J]. Epigenetics, 2012, 7: 1413–1420. DOI:10.4161/epi.22713 |
| [52] | Yuan L, Sheng X, Clark LH, et al. Glutaminase inhibitor compound 968 inhibits cell proliferation and sensitizes paclitaxel in ovarian cancer[J]. Am J Transl Res, 2016, 8: 4265–4277. |
| [53] | Yan YS, Hui L, Zhang X, et al. Histone modifications in senescence-associated resistance to apoptosis by oxidative stress[J]. Redox Biol, 2013, 1: 8–16. DOI:10.1016/j.redox.2012.11.004 |
| [54] | Friday RE, Oliver R, Welbourne T, et al. Directing glutamine and glucose metabolism with troglitazone and EGCG limits MCF-7 cell growth: Proceedings of the 101st Annual Meeting of the American Association for Cancer Research[C]. Washington, 2010: 48. |
| [55] | Zhang J, Wang G, Mao Q, et al. Glutamate dehydrogenase (GDH) regulates bioenergetics and redox homeostasis in human glioma[J]. Oncotarget, 2016, 295: 799–800. |
| [56] | Korangath P, Teo WW, Sadik H, et al. Targeting glutamine metabolism in breast cancer with aminooxyacetate[J]. Clin Cancer Res, 2015, 21: 3263–3273. DOI:10.1158/1078-0432.CCR-14-1200 |
| [57] | Papathanassiu AE, Hong AV. Inhibition of BCAT1 suppresses the expression of pro-metastatic proteins and reduces cancer metastasis: proceedings of the 105th Annual Meeting of the American Association for Cancer Research[C]. San Diego: AACR, 2014: 2683. |
| [58] | Mayers JR, Torrence ME, Danai LV, et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers[J]. Science, 2016, 353: 1161–1165. DOI:10.1126/science.aaf5171 |
| [59] | Esslinger CS, Cybulski KA, Rhoderick JF. Nγ-Aryl glutamine analogues as probes of the ASCT2 neutral amino acid transporter binding site[J]. Bioorg Med Chem, 2005, 13: 1111–1118. DOI:10.1016/j.bmc.2004.11.028 |
| [60] | Oppedisano F, Catto M, Koutentis PA, et al. Inactivation of the glutamine/amino acid transporter ASCT2 by 1, 2, 3-dithiazoles: proteoliposomes as a tool to gain insights in the molecular mechanism of action and of antitumor activity[J]. Toxicol Appl Pharmacol, 2012, 265: 93–102. DOI:10.1016/j.taap.2012.09.011 |
| [61] | Schulte ML, Khodadadi AB, Cuthbertson ML, et al. 2-Amino-4-bis(aryloxybenzyl)aminobutanoic acids: a novel scaffold for inhibition of ASCT2-mediated glutamine transport[J]. Bioorg Med Chem Lett, 2016, 26: 1044–1047. DOI:10.1016/j.bmcl.2015.12.031 |
| [62] | Brer A, Fairweather S, Brer S. Disruption of amino acid homeostasis by novel ASCT2 inhibitors involves multiple targets[J]. Front Pharmacol, 2018, 9: 785. DOI:10.3389/fphar.2018.00785 |
| [63] | Shanware NP, Mullen AR, Deberardinis RJ, et al. Glutamine: pleiotropic roles in tumor growth and stress resistance[J]. J Mol Med, 2011, 89: 229–236. DOI:10.1007/s00109-011-0731-9 |
| [64] | Speyer CL, Nassar MA, Hachem AH, et al. Riluzole mediates anti-tumor properties in breast cancer cells independent of metabotropic glutamate receptor-1[J]. Breast Cancer Res Treat, 2016, 157: 217–228. DOI:10.1007/s10549-016-3816-x |
| [65] | Possemato R, Marks KM, Shaul YD, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer[J]. Nature, 2011, 476: 346–350. DOI:10.1038/nature10350 |
| [66] | Denicola GM, Chen PH, Mullarky E, et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer[J]. Nat Genet, 2015, 47: 1475–1481. DOI:10.1038/ng.3421 |
| [67] | Pollari S, Käkönen SM, Edgren H, et al. Enhanced serine production by bone metastatic breast cancer cells stimulates osteoclastogenesis[J]. Breast Cancer Res Treat, 2011, 125: 421–430. DOI:10.1007/s10549-010-0848-5 |
| [68] | Maddocks ODK, Berkers CR, Mason SM, et al. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells[J]. Nature, 2013, 493: 542–546. |
| [69] | Yang M, Vousden KH. Serine and one-carbon metabolism in cancer[J]. Nat Rev Cancer, 2016, 16: 650–652. DOI:10.1038/nrc.2016.81 |
| [70] | Nilsson LM, Forshell TZP, Rimpi S, et al. Mouse genetics suggests cell-context dependency for Myc-regulated metabolic enzymes during tumorigenesis[J]. PLoS Genet, 2012, 8: e1002573. DOI:10.1371/journal.pgen.1002573 |
| [71] | Nilsson R, Jain M, Madhusudhan N, et al. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer[J]. Nat Commun, 2014, 5: 3128. DOI:10.1038/ncomms4128 |
| [72] | Zhang WC, Shyh-Chang N, Yang H, et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis[J]. Cell, 2012, 148: 259–272. DOI:10.1016/j.cell.2011.11.050 |
| [73] | Pacold ME, Brimacombe KR, Chan SH, et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate[J]. Nat Chem Biol, 2016, 12: 452–458. DOI:10.1038/nchembio.2070 |
| [74] | Mullarky E, Lucki NC, Zavareh RB, et al. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers[J]. Proc Natl Acad Sci U S A, 2016, 113: 1778–1783. DOI:10.1073/pnas.1521548113 |
| [75] | Vacchelli E, Aranda F, Eggermont A, et al. Trial watch: IDO inhibitors in cancer therapy[J]. Oncoimmunology, 2014, 3: e957994. DOI:10.4161/21624011.2014.957994 |
| [76] | Miller CL, Llenos IC, Dulay JR, et al. Upregulation of the initiating step of the kynurenine pathway in postmortem anterior cingulate cortex from individuals with schizophrenia and bipolar disorder[J]. Brain Res, 2006, 1073: 25–37. |
| [77] | Pilotte L, Larrieu P, Stroobant V, et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2, 3-dioxygenase[J]. Proc Natl Acad Sci U S A, 2012, 109: 2497–2502. DOI:10.1073/pnas.1113873109 |
| [78] | Yamamoto S, Hayaishi O. Tryptophan pyrrolase of rabbit intestine. D- and L-tryptophan-cleaving enzyme or enzymes[J]. J Biol Chem, 1967, 242: 5260–5266. |
| [79] | Cady SG, Sono M. 1-Methyl-DL-tryptophan, beta-(3-benzofuranyl)-DL-alanine (the oxygen analog of tryptophan), and beta-[3-benzo(b)thienyl]-DL-alanine (the sulfur analog of tryptophan) are competitive inhibitors for indoleamine 2, 3-dioxygenase[J]. Arch Biochem Biophys, 1991, 291: 326–333. DOI:10.1016/0003-9861(91)90142-6 |
| [80] | Kudo Y, Boyd CAR. The role of L‐tryptophan transport in L‐tryptophan degradation by indoleamine 2, 3‐dioxygenase in human placental explants[J]. J Physiol, 2010, 531: 417–423. |
| [81] | Metz R, Rust S, Duhadaway JB, et al. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: a novel IDO effector pathway targeted by D-1-methyl-tryptophan[J]. Oncoimmunology, 2012, 1: 1460–1468. DOI:10.4161/onci.21716 |
| [82] | Yue EW, Douty B, Wayland B, et al. Discovery of potent competitive inhibitors of indoleamine 2, 3-dioxygenase with in vivo pharmacodynamic activity and efficacy in a mouse melanoma model[J]. J Med Chem, 2009, 52: 7364–7367. DOI:10.1021/jm900518f |
| [83] | Austin CJD, Rendina LM. Targeting key dioxygenases in tryptophan-kynurenine metabolism for immunomodulation and cancer chemotherapy[J]. Drug Discov Today, 2015, 20: 609–617. DOI:10.1016/j.drudis.2014.11.007 |
| [84] | Rhrig UF, Majjigapu SR, Vogel P, et al. Challenges in the discovery of indoleamine 2, 3-dioxygenase 1 (IDO1) inhibitors[J]. J Med Chem, 2015, 58: 9421–9437. DOI:10.1021/acs.jmedchem.5b00326 |
| [85] | Zhang X, Sun XX, Xue D, et al. Conformation-dependent scFv antibodies specifically recognize the oligomers assembled from various amyloids and show colocalization of amyloid fibrils with oligomers in patients with amyloidoses[J]. BBA-Proteins Proteomics, 2011, 1814: 1703–1712. DOI:10.1016/j.bbapap.2011.09.005 |
| [86] | Mautino MR, Jaipuri FA, Waldo J, et al. NLG919, a novel indoleamine-2, 3-dioxygenase (IDO)-pathway inhibitor drug candidate for cancer therapy: Proceedings of the 104th Annual Meeting of the American Association for Cancer Research[C]. Washington: AACR, 2013: 491. |
| [87] | Nayak A, Hao Z, Sadek R, et al. A Phase I study of NLG919 for adult patients with recurrent advanced solid tumors[J]. J Immunother Cancer, 2014, 2(S3): P250. |
| [88] | Siu LL. BMS-986205, an optimized indoleamine 2, 3-dioxygenase 1 (IDO1) inhibitor, is well tolerated with potent pharmacodynamic (PD) activity, alone and in combination with nivolumab (nivo) in advanced cancers in a phase 1/2A trial: Proceedings of the American Association for Cancer Research Annual Meeting[C]. National Harbor, 2017: 1-5. |
| [89] | Salter M, Hazelwood R, Pogson CI, et al. The effects of a novel and selective inhibitor of tryptophan 2, 3-dioxygenase on tryptophan and serotonin metabolism in the rat[J]. Biochem Pharmacol, 1995, 49: 1435–1442. DOI:10.1016/0006-2952(95)00006-L |
| [90] | Dolušić E, Larrieu P, Moineaux L, et al. Tryptophan 2, 3-dioxygenase (TDO) inhibitors. 3-(2-(pyridyl) ethenyl) indoles as potential anticancer immunomodulators[J]. J Med Chem, 2011, 54: 5320–5334. DOI:10.1021/jm2006782 |
| [91] | Bakmiwewa SM, Fatokun AA, Tran A, et al. Identification of selective inhibitors of indoleamine 2, 3-dioxygenase 2[J]. Bioorg Med Chem Lett, 2012, 22: 7641–7646. DOI:10.1016/j.bmcl.2012.10.010 |
| [92] | Aitken JB, Austin CJD, Hunt NH, et al. The Fe-heme structure of met-indoleamine 2, 3-dioxygenase-2 determined by X-ray absorption fine structure[J]. Biochem Biophys Res Commun, 2014, 450: 25–29. DOI:10.1016/j.bbrc.2014.05.054 |
| [93] | Löb S, Königsrainer A, Zieker D, et al. IDO1 and IDO2 are expressed in human tumors: levo- but not dextro-1-methyl tryptophan inhibits tryptophan catabolism[J]. Cancer Immunol Immunother, 2009, 58: 153–157. DOI:10.1007/s00262-008-0513-6 |
| [94] | Sørensen RB, Køllgaard T, Andersen RS, et al. Spontaneous cytotoxic T-cell reactivity against indoleamine 2, 3-dioxygenase-2[J]. Cancer Res, 2011, 71: 2038–2044. DOI:10.1158/0008-5472.CAN-10-3403 |
| [95] | Zhang W, Ramamoorthy Y, Kilicarslan T, et al. Inhibition of cytochromes P450 by antifungal imidazole derivatives[J]. Drug Metab Dispos, 2002, 30: 314–318. DOI:10.1124/dmd.30.3.314 |
| [96] | Röhrig UF, Majjigapu SR, Chambon M, et al. Detailed analysis and follow-up studies of a high-throughput screening for indoleamine 2, 3-dioxygenase 1 (IDO1) inhibitors[J]. Eur J Med Chem, 2014, 84: 284–301. DOI:10.1016/j.ejmech.2014.06.078 |
| [97] | Daniele C, Saverio T, Ovidio B, et al. Expanding targets for a metabolic therapy of cancer: L-asparaginase[J]. Recent Patents Anti-Cancer Drug Discov, 2012, 7: 4–13. |
| [98] | Maggi M, Mittelman SD, Parmentier JH, et al. A protease-resistant Escherichia coli asparaginase with outstanding stability and enhanced anti-leukaemic activity in vitro[J]. Sci Rep, 2017, 7: 14479. DOI:10.1038/s41598-017-15075-4 |
| [99] | Phillips MM, Sheaff MT, Szlosarek PW. Targeting arginine-dependent cancers with arginine-degrading enzymes: opportunities and challenges[J]. Cancer Res Treat, 2013, 45: 251–262. DOI:10.4143/crt.2013.45.4.251 |
| [100] | Sica A, Porta C, Morlacchi S, et al. Origin and functions of tumor-associated myeloid cells (TAMCs)[J]. Cancer Microenviron, 2012, 5: 133–149. DOI:10.1007/s12307-011-0091-6 |
| [101] | Geiger R, Rieckmann JC, Wolf T, et al. L-Arginine modulates T cell metabolism and enhances survival and anti-tumor activity[J]. Cell, 2016, 167: 829–842. DOI:10.1016/j.cell.2016.09.031 |
| [102] | Fallarino F, Grohmann U, Vacca C, et al. T cell apoptosis by tryptophan catabolism[J]. Cell Death Differ, 2002, 9: 1069–1077. DOI:10.1038/sj.cdd.4401073 |
| [103] | Fallarino F, Grohmann U, You S, et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor ζ-chain and induce a regulatory phenotype in naive T cells[J]. J Immunol, 2006, 176: 6752–6761. DOI:10.4049/jimmunol.176.11.6752 |
| [104] | Kung HN, Marks JR, Chi JT. Glutamine synthetase is a genetic determinant of cell type-specific glutamine independence in breast epithelia[J]. PLoS Genet, 2011, 7: e1002229. DOI:10.1371/journal.pgen.1002229 |
| [105] | Souba WW. Glutamine and cancer[J]. Ann Surg, 1993, 218: 715–728. DOI:10.1097/00000658-199312000-00004 |
| [106] | Basun H, Forssell LG, Almkvist O, et al. Amino acid concentrations in cerebrospinal fluid and plasma in Alzheimer's disease and healthy control subjects[J]. J Neural Transm Park Dis Dement Sect, 1990, 2: 295–304. DOI:10.1007/BF02252924 |