 2018, Vol. 53
2018, Vol. 53

编者按
新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等多维性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
在未知NS5A蛋白三维结构和作用机制的情况下, 研制出首创的口服抗HCV药物达卡他韦(daclatasvir), 是运用化学和药物化学原理的成功范例。从化学基因组学概念出发, 筛选发现苗头, 通过观察与化学研究, 形成了对靶标和配体二聚体的认识, 在优化物化性质、双靶标活性和消除潜在毒性等多维度研究中, 多次运用骨架迁越和药效团替换, 以及广泛探索的构效关系, 达卡他韦的研制不失为教科书式的一例。
丙型肝炎病毒(HCV)感染由于无法用疫苗预防, 对既有的药物又容易产生耐药, 因而成为患者预防与治疗的难题。本研究项目开始之前, 业界多集中于研制蛋白酶和RNA聚合酶抑制剂, 百事美施贵宝公司(BMS)考虑另辟蹊径, 从其他的环节或靶标研制新的抗HCV药物, 以便联合用药, 为疗愈丙肝患者提供新的手段。
1.1 干预HCV的蛋白NS5AHCV是含有9.6 kb基因组的RNA病毒, 基因组编码的多聚蛋白有4种结构蛋白和6种非结构蛋白, 这些蛋白在HCV进入宿主细胞、复制和出芽等过程发挥不同的作用。非结构蛋白中的NS2和NS3具有蛋白酶活性, 催化病毒蛋白的成熟; NS4A是NS3的辅酶; NS4B催化RNA的合成和诱导宿主细胞膜的改变。以这些蛋白作为靶标已有成功的药物上市。非结构蛋白NS5A是含有447个氨基酸残基组成的结合锌离子的磷蛋白, 在HCV病毒复制、装配和调节宿主细胞功能以保持慢性感染状态等过程起关键作用, 即使没有酶催化功能, 这些功能也成为研制药物的靶标(Lemon SM, McKeating JA, Pietschmann T, et al. Development of novel therapies for hepatitis C. Antiviral Res, 2010, 86: 79-92)。BMS公司以此为研制抗丙肝药物的目标。
1.2 化学基因组学用于细胞模型筛选——评价活性和毒性生物学上的基因筛选旨在确定蛋白质功能, 是将细胞随机地发生变异, 通过筛选生成的突变体所呈现的重要特征, 确定特定基因发生的对应突变。新药研究中应用化学基因组学(chemical genetics)类似于生物学的基因筛选, 是用细胞筛选小分子化合物库, 通过产生重要的表型(phenotype)变化, 鉴别相应的靶标蛋白, 既寻找苗头或先导物, 也确定靶标的功能(Stockwell BR. Chemical genetics: ligand- based discovery of gene function. Nat Rev Genet, 2000, 1: 116-125)。
病毒只能在宿主细胞内复制, 理论上讲, 抑制病毒的复制由于只干扰被感染细胞的特有病毒蛋白, 不会影响正常细胞, 但由于化合物或生物大分子的杂泛性(promiscuity), 也可能对正常细胞产生影响, 所以, 目标化合物不应干扰宿主蛋白, 以避免毒副作用。
为此, BMS用3种细胞模型分别评价化合物活性、选择性和细胞毒性, 即用HCV NS5A的1b基因亚型复制子感染的人肝细胞评价对NS5A活性(GT-1b EC50); 用小牛病毒性腹泻病毒(BVDVEC50)感染细胞评价化合物的选择性; 还评价化合物的细胞毒作用(CC50)。
2 苗头化合物和先导物演化 2.1 苗头化合物的发现BMS公司用高通量筛选方法筛选了一百万个化合物, 从中发现一个具有初步活性和选择性的噻唑烷酮化合物(1, GT-1b EC50 0.58 μmol·L-1, BVDV EC50 > 50 μmol·L-1, CC50 > 100 μmol·L-1)。但该苗头分子具有明显缺点:分子量较大(MW = 587), 疏水性过强(clogP > 5), 有较大的极性表面积(PAS = 139 2), 以及噻唑烷酮手性碳的互变异构造成的不稳定性等。因而需要结构改造, 演化成先导物(hit-to-lead) (Bell TW. Drugs for hepatitis C: unlocking a new mechanism of action. ChemMedChem, 2010, 5: 1663-1665)。
|
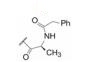
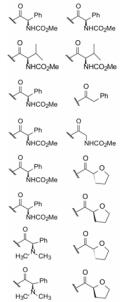
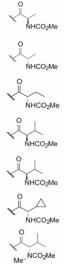
变换化合物1的结构, 母核5-苯基噻唑烷酮所连接的呋喃环、氟苯基亚胺和氨基酸片段都是可变换的部分。首先, 将呋喃和氟苯基固定不变, 只变换S-丙氨酸片段, 表 1列出了合成的有代表性的化合物1~6。
| Table 1 Structures and activity of compounds with varied amino acids |

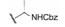
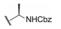
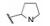
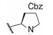

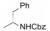
表 1中(以及未列出的)化合物构效关系提示, 手性Cα的S构型(氨基酸的L构型)活性强于相应的R构型(D构型), 大约高1~2个数量级, 提示天然氨基酸有利于活性, 而且Cα上以连接小体积基团为宜。
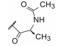
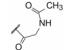
进而考察L-丙氨酸和L-脯氨酸的酰基对活性影响, 表 2的构效关系提示, 将苄氧羰基换成苯甲酰基(7和8)或苯丙酰基(11和12)都使活性降低, 而苯乙酰基化合物9和10的活性提高大约100倍, 9是活性最强的化合物。
| Table 2 Effect of varied acyl groups on the activity |
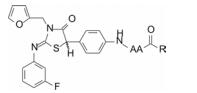
变换呋喃和氟苯片段的构效关系提示, 呋喃环换成吡啶或吡嗪等极性较强的芳环, 显著提高活性, 间氟苯基环上若引入高脂溶性基团则降低活性(数据省略) (Romine JL, Laurent DRS, Leet JE, et al. Inhibitorsof HCV NS5A: from iminothiazolidinones to symmetrical stilbenes. ASC Med Chem Lett, 2011, 2: 224-229)。
3 活性二聚化合物 3.1 噻唑烷酮化学不稳定性的意外发现深入研究化合物9, 发现9的DMSO溶液放置数天后, 分子发生氧化重排, 生成硫代乙内酰脲(13)以及降解产物硫脲(14)和酮酸(15)。重排和分解的历程推测如图 1所示。

|
Figure 1 Rearrangement process of thiazolinone into thiohydantoin or thiourea and ketoacid |
由这种不稳定性联想到抗病毒培养液是否也会发生变化, 结果发现化合物9在感染细胞培养液中也是不稳定的, 然而无论9的降解程度如何, 仍然对HCV复制子有强效抑制活性, 由这个意外发现推论可能是微量的降解产物(或混合物)具有抑制活性, 遂经HPLC分离出两个化合物, 抑制HCV复制子活性EC50分别为43 nmol·L-1和0.6 nmol·L-1, 其中一个是亚稳定体, 加热变成另一个热力学稳定物。经MS和NMR证明是9的二聚物16, MW = 1139, EC50为3 nmol·L-1, 具有很高活性。还发现16对HCV复制子突变体Y93H也有敏感的抑制活性, 而单体9对该变异株活性很弱(EC50 > 5 μmol·L-1)。化合物9生成二聚体的机制推测是在O2作用下拔除C5-H, 生成自由基(暂被硫原子和羰基稳定)偶联而成。
二聚体16对野生型和Y93H突变体都保持高抑制活性, 提示抑制剂的药效团呈二聚分布, 这为新分子的设计策略提供了新的依据。16成为里程碑式的发现(Lemm, JA, Leet JE, O’BoyleDR, Ⅱ, et al. Discovery of potent hepatitis C virus NS5A inhibitors with dimeric structures. Antimicrob Agents Chemother, 2011, 55: 3795-3802)。

|
活性二聚体的发现, 联想到靶标蛋白可能以二聚体的形式起作用, 后来实验证明HCV NS5A果然以二聚体形式与16相结合(Tellinghuisen TL, Mar cotrigiano J, Rice CM. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature, 2005, 435: 374-379)。基于药效团原理, 推论外端的基团应分别结合于靶标二聚体的结合位点上, 这时噻唑烷酮可能是起连接基的作用, 未必是参与结合的药效团因素。这个科学假定成为简化化合物16结构的切入点。
最简捷的方法是删除噻唑烷酮和连接的呋喃和氟苯片段。并将L-丙氨酸换成L-脯氨酸(分子简化后可适当增添原子, 并且前已证明L-脯氨酸也是高活性片段), 得到以二苯乙烷为骨架的化合物17 (GT-1b E50 30 nmol·L-1), 相对分子质量减少到642。17显示出较高的活性。为降低二苯乙烷骨架的柔性(二聚体16为刚性分子), 合成了二苯乙烯化合物18 (E50 GT-1a > 10 μmol·L-1; GT-1b 0.086 nmol·L-1), 对GT-1b的抑制活性显著提高。
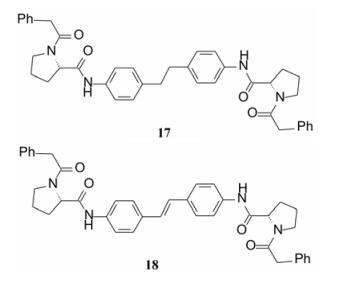
|
为了考察化合物抑制靶标是否需要对称地结合于相同对应的位点, 以18为模板化合物, 固定一侧结构, 变换另一侧的L-脯氨酸的苯乙酰基或变换脯氨酸片段, 合成的化合物及其活性列于表 3中。结果表明, 苯乙酰基被丙酰基替换, 化合物19活性降低200倍, 无酰基的20和没有脯氨酸片段的21活性降低到微摩尔级, R为H的游离氨基(22)失去活性。脯氨酸换成丙氨酸片段(23), 活性降低10倍, 23的苯乙酰基换成乙酰基(24)以及去掉Cα-甲基(25)都使活性显著下降。这一组化合物的构效关系表明: ①二聚型分子的对称性似乎是必要的(后来用其他骨架证明只要在适宜距离有类似的药效团, 非对称分子也可呈现高活性); ②脯氨酸片段是优选的; ③氮原子须经酰基, 酰基尺寸不宜太大等。
| Table 3 Activity of asymmetric compounds with stilbene skeleton |
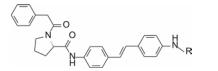

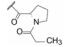
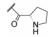
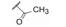
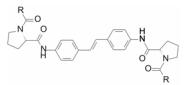
进而对脯氨酸片段的四氢吡咯环作扩环、并环或取代等变换, 活性仍以化合物18为最佳(St. Laurent DR, BelemaM, Gao M, et al. HCV NS5A replication complex inhibitors. Part 2: investigation of stilbene prolinamides. Bioorg Med Chem Lett, 2012, 22: 6063- 6066)。
3.4 双重抑制剂的研究为了提高抗HCV效果, 要求对GT-1a和GT-1b复制子有双重作用, 即泛抑制活性。化合物18虽然对GT-1b有较高活性, 但对GT-1a未见活性(EC50 > 10 μmol·L-1)。为了研制有双重作用的泛抑制剂, 以对称性二苯乙烯分子18为起始物, 系列地改变脯氨酸酰化基团, 希望在改变外端的基团或片段中呈现并提高双抑制活性。发现化合物26对GT-1a呈现弱活性(EC50 = 0.95 μmol·L-1), 但对GT-1b的活性损失较大。将乙基苯环置换为乙基吡啶(27), 氮原子的引入提高了对GT-1b活性。此外, 通过比较18与26的最佳构象的苯环与酰基的两面角(分别为-114.6 和-116.6 ), 相差甚小, 推论活性差异不是因乙基的位阻所引起的构象不同所致, 可能是增加了疏水性的缘故。
进而将乙基吡啶增加两个碳原子, 环合, 芳化, 变成异喹啉28, 此时对两种复制子的抑制活性都有提高, 可能是增强了π-π相互作用的缘故。由于萘甲酰的活性很弱, 提示氮原子起重要作用。以化合物28为起点作不同的取代, 合成了多种取代的异喹啉类似物, 优选出化合物29 (表 4)。
| Table 4 Activity on dual targets of stilbene compounds |
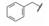
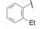
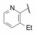
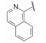
与此同时, 还研究了二聚物之间不同的连接基对活性的影响。由于反式二苯乙烯化合物在DMSO溶液中可发生trans-cis异构化, 因而探索用其他稳定的共轭连接基, 如乙炔或芳杂环等。合成有代表性的化合物是外端的脯氨酸片段被N-苯乙酰化和3-氯-5-甲氧基异喹啉甲酰化(因化合物29显示高活性), 结果列于表 5中。虽然成对的化合物都显示出异喹啉系列的双重抑制活性较高, 但因为有细胞毒作用, 中止了异喹啉结构类型的研究(Laurent DRS, Serrano-Wu MH, Belema M, et al. HCV NS5A replication complex inhibitors. Part 4. Optimization for genotype 1a replicon inhibitory activity. J Med Chem, 2014, 57: 1976-1994)。
| Table 5 Activity of dimer compounds with varied linkers |
二苯乙炔骨架的化学稳定性强于二苯乙烯骨架。为了优化这类二聚物的活性, 将前述的构效关系用于二苯乙炔系列的设计上(这种“代入法”有时是成功的)。例如在苯乙酰的α碳上合成含有碱性基团取代的化合物, 评价对GT-1a和GT-1b的抑制活性, 列于表 6中。构效关系表明, 该系列呈现双重抗HCV作用, 对GT-1b的活性强于GT-1a, 手性α碳为R构型的活性高于相应的S构型。其中37和42都有强效的抑制GT-1b活性, 但是对GT-1a的抑制作用较弱, 为了使两种复制子的抑制活性匹配, 需要对拓扑结构作优化调整(Belema M, Nguyen VN, Laurent DRS, et al. HCV NS5A replication complex inhibitors. Part 5: discovery of potent and pan-genotypic glycinamide cap derivatives. Bioorg Med Chem Lett, 2013, 23: 4428- 4435)。
| Table 6 Activity of diphenylacetylene compounds with basic moiety. *The configuration of carbon atom with R group in compounds 40-47 is R-form |
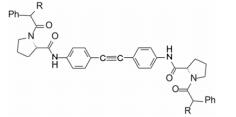



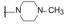
虽然二苯乙炔骨架的稳定性强于二苯乙烯, 但对于GT-1a和GT-1b病毒复制子的双重抑制活性强度差异较大, 抑制GT-1a作用较弱, 因而需做进一步优化。另外, 这类化合物是酰化的二苯胺乙炔, 在体内有可能代谢水解生成二苯胺衍生物, 会引起基因突变和细胞毒性, 呈现特质性药物毒性(idiosyncratic drug toxicity, IDT)。这个问题可通过骨架迁越加以克服。
药物分子设计中认为酰化苯胺片段与苯并咪唑之间存在有相似的结构和几何特征, 可视作广义的电子等排, 因而可以互换。为此合成了以二苯并咪唑乙炔为骨架的分子, 有代表性的化合物列于表 7。
| Table 7 Activity of bis-benzimidale (-benzoxazole) acetylene compounds |

表 7的数据提示, 将酰基苯胺环合成苯并咪唑, 48和49保持了抑制GT1b活性, 但对GT-1a活性降低了数10倍, 而苯并噁唑化合物50和51对GT-1a未呈现活性。所以, 苯并咪唑骨架本身不值得深入研究, 但提供了进一步变换结构的思路。
4.2 二聚物外端药效团的距离对活性影响乙炔键用噁唑环替换作为骨架, 合成的化合物通过改变取代基的位置研究构效关系, 发现由于位置异构使外端药效团之间的距离发生改变, 引起活性的变化。表 8列出有代表性的化合物。54的胺酰基由对位移至间位, 化合物55抑制GT-1b的活性变化不大, 而对GT-1a活性提高了70倍。52、56和57三个化合物互为区域异构体, 只有57 (m-, p-)对GT-1a呈现高活性。这些数据说明变换骨架和连接的位置以调整外端药效团的空间距离, 可以得到对GT-1a和GT-1b都有高活性的双重抑制剂。
| Table 8 Activity of compounds with varied scaffold |

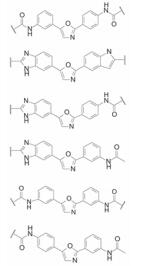
综合前述的具有双重抑制活性的化合物, 说明骨架允许多种变换, 例如双键、炔键或杂环等, 它们的共同特征是具有共轭结构, 外端药效团的空间距离对双重抑制活性敏感等。
由于二苯并咪唑乙炔或由杂环相连的分子对GT-1a的抑制活性不高(例如48、49和53等), 为了提高对GT-1a活性, 骨架变换的策略是将苯并咪唑拆解成苯基咪唑, 即由稠合环变成单键连接的苯环和咪唑环, 这显然增加了骨架的长度, 需要去除中间的三键或杂芳环片段, 维持端基间的距离, 从而形成了二咪唑联苯的结构。
4.3.1 以二咪唑联苯-苯甘氨酰四氢吡咯为骨架的对称和非对称化合物以二咪唑联苯为骨架, 经四氢吡咯片段, 合成了N-苯甘氨酰化合物58, 对GT-1a和GT-1b均呈现高活性, 对于牛病毒性腹泻病毒(BVDV)的活性很弱, 提示选择性作用强。然而58的溶解性差, 大鼠灌胃的吸收性很低, 可能是相对分子质量大的缘故。将苯甘氨酸变换成非天然的缬氨酸(保持α碳的R构型, 此时选取R构型只是效仿了以前系列的R强于S构型的构效关系, 实际上S构型活性更强), 得到的化合物59活性降低数万倍。即使保留单边的苯甘氨酸片段, 如化合物60~63虽然仍有抑制GT-1b活性, 但对GT-1a活性降低了400~10 000倍。然而单侧换成苯乙胺片段, 并简化另侧结构, 化合物64和65提高了双抑制活性。表 9列出了有代表性的二咪唑联苯类化合物的活性。
| Table 9 Activity of compounds with bis-imidazolyl biphenyl scaffold |
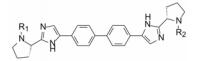
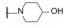
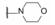
在3.6节合成的二苯基乙炔系列含有碱性胺类化合物, 其中37和42有较强的双抑制活性, 由于化合物64和65有良好的活性和增强的溶解性, 作为新一轮先导物, 合成了对称性的含有碱性氮原子的化合物。表 10中化合物的构效关系表明, 含胺片段的尺寸对活性影响不大, 但碱性的强弱则与活性相关, 碱性低的化合物如含有吗啉和氟代四氢吡咯的70和71抑制GT-1a的活性明显减弱。
| Table 10 Activity of compounds with bis-imidazolyl biphenyl scaffold and basic moiety |
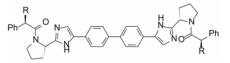
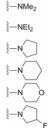
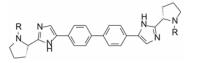
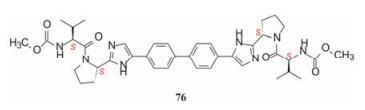
在4.3.1节叙述了苯基甘氨酸(58)的R构型的活性显著高于非天然缬氨酸类似物(59), 这与相同的构型有相似活性的药物化学常见现象有所不同。为此, 考察了不同构型和基团对活性的影响。表 11列出了代表性化合物的构效关系。合成的含有D-和L-丙氨酸(甘氨酸的α碳分别是R-和S-构型)片段是一对差向异构体(分别为72和73)对GT-1a和GT-1b的活性相差很大, 高活性化合物73的α碳构型与前述高活性的苯基甘氨酸58 (表 9)的构型相反, 而与低活性的化合物46 (非天然的缬氨酸)的构型一致, 反映出该结构区域对活性的敏感性。从而合成了天然缬氨酸化合物76, 76的活性非常高, 抑制GT-1a和GT-1b的EC50值分别是0.050和0.009 nmol·L-1, 它的对映体75的活性非凡的弱。化合物77是将76的异丙基换成环丙基, 虽具有相同构型, 活性显著减弱, 或许是环丙基具有不饱和因素的缘故。化合物78是N-甲基化的76, 活性很低, 推测缺少N-H氢键给体的因素。
| Table 11 Activity of compounds with symmetric α-alkyl glycine series |
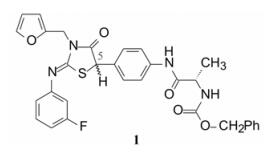
在结构骨架和外端药效团的变换中, 各个阶段出现对GT-1a和GT-1b复制子有双重活性的里程碑式的数个化合物, 分别用大鼠、犬和食蟹猴等动物评价药代动力学性质, 主要包括口服(灌胃)生物利用度, 不同剂量的血浆AUC, 和化合物灌胃后24 h的血浆暴露量等, 其中化合物76的药代动力学性质最佳, 列于表 12。也由于对GT-1a和GT-1b复制子有高抑制活性, 遂确定为候选化合物进入全面开发阶段, 定名为达卡他韦(daclatasvir) (Belema M, Lopez OD, Bender JA, et al. Discovery and development of hepatitis C virus NS5A replication complex inhibitors. J Med Chem, 2014, 57: 1643-1672. Belema M, Meanwell NA. Discovery of daclatasvir, a pan-genotypic hepatitis C virus NS5A. J Med Chem, 2014, 57: 5057-5071)。
|
| Table 12 The pharmacokinetic parameters of compound 76 in various animals |
经临床研究达卡他韦为有效的抗丙肝病毒药物, 于2015年经FDA批准上市, 成为第一个以HCV NS5A蛋白为靶标的口服抗丙肝病毒药物。与聚乙二醇干扰素和利巴韦林合用或与其他直接作用于病毒的药物如阿那匹韦或索非布韦联合用药可达到良好的治疗效果。
6 与NS5A蛋白的结合模式根据X-射线分析的GT-1b的25~215残基所构成的结构域Ⅰ以及由NMR分析的两亲性螺旋域构成的结构, 经同源模建方法构建了二聚体, 用化合物66经分子对接得到了复合物的三维结构, 如图 2所示。化合物66与达卡他韦的结构骨架相同, 结合模式应是相似的。作为线型分子, 66对称地跨越结合于二聚体表面, 联苯基与Phe37和Pro58发生疏水-疏水相互作用。咪唑的氮原子与Gln62侧链形成氢键结合。四氢吡咯和苯环分别同Leu28和Pro29发生疏水-疏水相互作用(O’Boyle DR Ⅱ, Sun JH, Nower PT, et al. Characterizations of HCV NS5A replication complex inhibitors. Virology, 2013, 444: 343-354)。

|
Figure 2 (a) The binding model of compound 66 with NS5A dimer; (b) Diagrammatic sketch of interaction of compound 53 with amino acid residues of NS5A |
在靶标的三维结构不明确或作用机制未经阐明的情况下, 药物分子设计大都靠分析构效关系指导研发方向, 有人比作摸着石头过河, 其实远不如此。摸着石头每前进一步趋近彼岸, 因为目标明确; 而借助构效关系的反馈和迭代以优化化合物, 有时未必如愿, 因而更像是“试错” (trial and error), 在排除大量的误导和“伪证”后, 获得真知。这就是新药创制长周期高风险之所在。
本文从药物化学视角简要地分析了达卡他韦从发现苗头到成功上市, 折射出在研发的路径上更多的是“试错”。下面梳理一些重要的发现与节点, 或许有益于借鉴与思考(图 3)。

|
Figure 3 Summary of investigation path of daclatasvir |