 2018, Vol. 53
2018, Vol. 53



血根碱(sanguinarine, 1), 化学名为2:3, 7:8-双亚甲二氧基-5-甲基苯并[c]菲啶-5-阳离子季铵盐, 是一种天然的苯并[c]菲啶型生物碱。根据文献, 血根碱存在于多个科属的植物中[1], 其存在形式包括季铵型、5, 6-二氢型叔胺结构及其6-取代类似物; 并通常与多种同类型或类似型生物碱共存; 如罂粟科博落回属植物博落回[2, 3]、小果博落回[4]和Bocconia integrifolia[5]等均含有丰富的二氢血根碱及其6-取代衍生物; 6-甲氧基二氢血根碱经季铵化处理可以得到血根碱[2];白屈菜属植物白屈菜中也含有少量的二氢血根碱[6]。采用HPLC方法还从白屈菜中鉴定了血根碱, 其含量为0.003% [7]。从罂粟科植物木罂粟Bocconia frutescens中也分离得到了血根碱[8]。血根碱具有抗菌、抗病毒、抑制胆碱乙酰转移酶[1]、抗炎、杀虫、改善肝功能[9]、抗高血压[8, 10]、肿瘤细胞毒性和抗肿瘤等广泛的生理作用[1, 11], 在包括医药、兽药和农药等多个领域均受到研究人员的关注[9];但其同时也具有在多种常见溶剂中溶解性能较差的性质, 不利于对其进行药理研究和包括在农药乳剂制备在内的有关方面的实际应用。文献报道, 苯并[c]菲啶型生物碱的季铵盐结构是影响其生物活性的重要因素之一, 而以季铵盐结构为存在形式, 在一定范围内改善其溶解性有助于改善其生物活性; 因此, 采用各种手段改善季铵盐结构的溶解性、提高生物活性的研究也受到了研究人员的关注[12]。此外, 研究发现, 在生理的水溶液条件下, 苯并[c]菲啶型生物碱以活性的季铵盐结构与非活性的6-羟基二氢型叔胺结构存在平衡关系, 而改善季铵盐结构的溶解性是促使此平衡关系向季铵盐结构移动的有效方法[12, 13]。因此, 开展血根碱的药物化学研究, 从包括亲水和亲脂的广义的概念上改善溶解度以及提高生物活性强度的角度对血根碱进行结构修饰, 是一个值得尝试的研究课题。本文报道采用汇聚合成的策略合成烃基-去-血根碱- N5-甲基衍生物的研究。显然, 通过用不同的亲脂性更强的烃基取代-N5-甲基有助于改善目标化合物的脂溶性。同时, 本文还报道对目标化合物的生物活性评价以及构效关系分析。
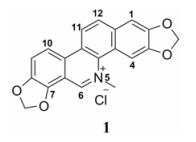
|
天然存在的苯并[c]菲啶型生物碱的N5-取代基均为甲基, 至今未见从天然资源中分离得到烃基-去-血根碱-N5-甲基衍生物的文献报道; 从天然资源中直接获得具有系列性特征的烃基-去-血根碱-N 5-甲基衍生物更是没有可能性。本文采用汇聚合成的策略对烃基-去-血根碱-N 5-甲基衍生物进行全合成研究, 以获得具有系列性特征的目标化合物, 进行肿瘤细胞株生长抑制活性的评价和构效关系研究。在目标化合物的合成中, 作为关键中间体的去甲基血根碱的合成是主要的挑战性工作之一。通过查阅文献可知, 已经有数种合成苯并[c]菲啶衍生物的方法被报道[14-19], 其中包括采用自由基芳构化路线合成血根碱结构类似物硫氢酸化7-羟基-8-甲氧基-5-甲基-2, 3-亚甲二氧基苯并[c]菲啶-5-阳离子季铵盐二水合物(NK109)的文献报道[14]。特别值得指出的是, NK109具有很强的抗肿瘤活性, 曾进入临床研究[14, 20];与NK109一样, 血根碱的5, 6位亚胺型季铵盐正离子结构是其肿瘤细胞毒作用的关键位点, 将亚胺结构还原为叔胺结构会导致细胞毒作用的消失[13]。本文设计的烷基-去-血根碱-N 5-甲基衍生物不仅可以通过增加位阻作用对5, 6位亚胺型季铵盐正离子结构进行“保护”, 而且可以增加目标物的脂溶性, 促进前述的平衡关系移向活性的亚胺型季铵盐结构。
采用汇聚合成的方式合成目标化合物(图 1)。首先对去甲基血根碱进行全合成; 将起始原料6-溴- 2, 3-二羟基苯甲醛与二溴甲烷经常规的二氧-去-二溴亲核取代反应得到醛合成子6-溴-2, 3-亚甲二氧基苯甲醛(2); 以5-硝基-2, 3-萘二酚为起始原料, 与二溴甲烷反应, 经上述同样的过程, 得到5-硝基-2, 3-亚甲二氧基萘(3), 再经催化氢化反应得到氨合成子6, 7-亚甲二氧基萘-1-胺(4)。合成子2与4在酸催化下生成席夫碱(5), 再经二甲胺硼烷还原得到仲胺结构6, 6经三丁基锡烷和AIBN诱导的自由基关环反应生成去甲基血根碱(7) (合成路线1)。

|
Figure 1 Retrosynthetic analysis to the alkyl-de-(sanguinarine-N5-methyl) derivatives by converging synthesis |

|
Reagents and conditions: (ⅰ) Dibromomethane, K2CO3, CuO, DMF, 110 ℃; (ⅱ) Dibromomethane, K2CO3, CuO, DMF, 110 ℃; (ⅲ) H2/Pd, MeOH, rt; (ⅳ) CF3COOH, toluene, 80 ℃; (ⅴ) Dimethylamine-borane, acetic acid; (ⅵ) Bu3SnH, toluene, N2, AIBN, 110 ℃ Scheme1 Synthesis of 2, 3:7, 8-dimethylenedioxybenzo[c]phenanthridines. |

|
Scheme2 Syntheses of quaternary 2, 3:7, 8-dimethylenedioxy-5-alkylbenzo[c]phenanthridin-5-ium chlorides (9a-9f) |
在醛合成子2与胺合成子4反应生成席夫碱5的过程中, 以甲苯为溶剂, 在80 ℃条件下滴加催化量的三氟乙酸, 5 min内即析出大量沉淀, 并经检测确定为目标产物席夫碱; 这一过程显著快于文献报道的其他类似席夫碱的合成。在具有类似结构的席夫碱的合成中多使用相对剧烈的反应条件, 如在NK109的合成中, 以甲苯为溶剂回流反应达1.5 h[14]。其原因可以归结为, 与文献中类似席夫碱的甲氧基和羟基对比, 本文目标产物席夫碱中的两个亚甲二氧基显著降低了其在反应混合液中的溶解度, 使得生成的席夫碱从反应体系中析出, 加快了反应速度, 并促进平衡正向移动。
以所获得的去甲基血根碱为底物, 以不同的正烷基羧酸为溶剂和反应试剂, 在硼氢化钠条件下生成N-烷基取代5, 6-二氢-2:3, 7:8-双亚甲二氧基-5-甲基苯并[c]菲啶(8)。8在碱性条件下经DDQ氧化和盐酸盐化反应生成目标产物烷基-去-血根碱-N5-甲基衍生物, 即2, 3:7, 8-双亚甲二氧基-N5-烷基苯并[c]菲啶- 5-阳离子季铵盐(9)[19](合成路线2)。
根据对反应现象的观察, 在N5-烷基取代二氢型产物(8)的合成中, 随着脂肪链长度的增加, 反应活性明显下降, 产率降低; 表明对于脂肪链过长的结构, 该反应不适合大量生产。但相比于采用三氟甲磺酸甲酯或硫酸二甲酯等甲基化试剂进行甲基化反应而言, 本文方法具有反应条件温和、时间短、使用范围广的优点, 且所合成产物具有结构系列性特征; 因此, 适合快速合成化合物库进行活性筛选。
所有目标产物的结构均经1H NMR和13C NMR以及ESI-MS+进行了确证。在1H NMR图谱中, 2, 3:7, 8-双亚甲二氧基苯并[c]菲啶母核在δH 8.11~8.78显示出两组芳香型AB自旋-自旋偶合信号, 偶合常数为8.7~9.0 Hz, 归属为两个连-四取代的苯环上CH(9)= CH(10)和CH(11)=CH(12)的共振信号。在δH 7.75~10.23区域内的3个分别含有1个质子的芳香质子单峰信号归属于CH(1)、CH(4)和CH(6)的共振。在δH 6.60~6.33区域内的两个分别含有两个质子的单峰信号归属2, 3-和7, 8-位的两个亚甲二氧基的共振。1H NMR图谱中其余所有的信号均出现在高场区, 结合化学位移、偶合常数和积分值判断, 这些信号均与所合成目标化合物的N5位正烷基结构相一致。所有目标化合物的13C NMR图谱以及ESI-MS+数据也都与各个化合物的结构相一致(见实验部分)。
2 目标化合物的肿瘤细胞生长抑制活性评价采用MTT法[21]评价了所合成的烷基-去-血根碱-N5-甲基衍生物对人结肠癌细胞(HCT-8)、人肝癌细胞(Bel7402)、人宫颈癌细胞(HeLa)、人肺癌细胞(A5409)和人胃癌细胞(BGC-823)的生长抑制活性, 并与天然的血根碱结构进行了比较。实验结果表明, 各个目标化合物均显示出对肿瘤细胞株生长的抑制活性, 3次重复试验的平均活性数据IC50值在1×10-6~1×10-7 mol·L-1范围, 均优于阳性药五氟尿嘧啶(5-Fu)。排除可能存在的误差, 可以看出, 随着N-5烷基取代基碳链长度的增加, 活性也相应增加; 其中, 化合物9f抑制宫颈癌细胞HeLa生长的IC50值为阳性药的22倍, 为天然血根碱母核结构的2倍。结合文献报道, 以上结果进一步印证了5, 6位亚胺型季铵盐正离子结构及目标化合物脂溶性的改善对肿瘤细胞生长抑制活性的关键性作用, 即在5位N上进行结构修饰, 改变烷基侧链的长度, 不仅能增加位阻保护活性中心, 分散电荷增加衍生物脂溶性, 而且可以通过分子间作用力轻微扭曲分子形状, 使活性提高。但是, 化合物9b和9f的活性处于非常接近的同一数量级, 这可能与9b的溶解度适中有关, 因为, 烷基链太长, 可能会导致溶解度降低, 活性和毒性同时增加。因此, 本文为进一步的构效关系研究以及其他方面的活性评价提供了有效参考数据。
实验部分1H NMR和13C NMR分别用Varian Mercury-300型、400型或Bruker AV-III-500型核磁共振波谱仪测定, 溶剂为DMSO-d6 (D, 99.9%)或CDCl3 (D, 99.8%), TMS为内标。正离子模式ESI-MS用JEOL Accu TOF CS (JMS T100CS)型质谱仪测定。柱色谱硅胶(200~300目)和薄层色谱硅胶(GF254)均为青岛海洋化工厂产品。所用溶剂和试剂均为市售分析纯, 未经进一步处理直接使用。
1 化学合成 1.1 汇聚法合成去甲基血根碱 1.1.1 5-溴苯并[d][1, 3]二噁英-4-甲醛(2)的合成在100 mL三口瓶中将6-溴-2, 3-二羟基苯甲醛(1.85 g, 8.5 mmol)溶于30 mL DMF中, 加入碳酸钾(2.35 g, 17 mmol), 搅拌均匀后加入氧化铜粉末(0.1 g)和二溴甲烷(1.19 mL, 17 mmol), 在110 ℃下搅拌。经TLC (石油醚-乙酸乙酯5:1, v/v)监测反应完成后, 冷却至室温, 滤除固体碳酸钾和氧化铜, 将滤液倒入150 mL水中, 析出固体。过滤, 将滤饼用异丙醇重结晶得淡黄色粉末1.0 g, 收率51.3%。1H NMR (300 MHz, DMSO-d6) δ 10.11 (s, 1H), 7.18 (d, J = 8.2 Hz, 1H), 7.09 (d, J = 8.2 Hz, 1H), 6.21 (s, 2H)。
1.1.2 5-硝基萘并[2, 3-d][1, 3]二噁英(3)的合成在100 mL三口瓶中将5-硝基-2, 3-萘二酚(2.40 g, 11.7 mmol)溶于30 mL DMF中, 然后加入碳酸钾(3.3 g, 23.9 mmol), 搅拌均匀, 加入氧化铜粉末(0.2 g)和二溴甲烷(1.6 mL, 23.4 mmol), 在110 ℃下搅拌。经TLC (石油醚-乙酸乙酯5:1, v/v)监测反应完成后, 冷却到室温, 滤除固体碳酸钾和氧化铜。将滤液倒入150 mL水中, 析出黑色固体。过滤, 滤饼用异丙醇重结晶得黄色粉末1.40 g, 收率56.0%。1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 7.8 Hz, 1H), 7.97 (s, 1H), 7.90 (d, J = 8.1 Hz, 1H), 7.37 (dd, J = 7.8, 8.1 Hz, 1H), 7.19 (s, 1H), 6.13 (s, 2H)。
1.1.3 萘并[2, 3-d][1, 3]二噁英-5-氨(4)的合成在100 mL茄型瓶中将3 (1.0 g, 4.6 mmol)溶于10 mL甲醇中, 然后顺序加入1 mL醋酸和10%钯碳0.1 g、连接氢化装置、换气3次后通入氢气, 于室温下搅拌。经TLC (石油醚-乙酸乙酯5:1, v/v)监测反应完成后, 将钯碳滤除; 将滤液浓缩后得黑色粗品; 粗品经硅胶柱色谱纯化, 采用石油醚-乙酸乙酯10:1 (v/v)为洗脱剂进行洗脱, 得到褐色粉末0.73 g, 收率85.0%。1H NMR (300 MHz, DMSO-d6) δ 7.46 (s, 1H), 7.12 (s, 1H), 7.01 (dd, J = 7.9, 7.4 Hz, 1H), 6.92 (br d, J = 7.9 Hz, 1H), 6.54 (dd, J = 7.4, 1.1 Hz, 1H), 6.06 (s, 2H), 5.43 (s, 2H)。
1.1.4 N-[(5-溴苯并[d][1, 3]二噁英-4-基)亚甲基]萘并[2, 3-d][1, 3]二噁英-5-胺(5)的合成在250 mL茄型瓶中将2 (10.0 g, 43.7 mmol)和4 (8.2 g, 43.7 mmol)溶于150 mL甲苯中并加热至80 ℃, 然后滴加1滴三氟乙酸, 溶液瞬间变清澈, 之后析出亮黄色沉淀。将反应混合液继续搅拌5 min后冷却至室温并过滤, 将滤饼在室温下抽滤至干燥, 得黄色固体13.5 g, 收率77.6%。1H NMR (400 MHz, CDCl3) δ 8.84 (s, 1H), 7.69 (s, 1H), 7.56 (d, J = 8.0 Hz, 1H), 7.33 (dd, J = 8.0, 8.0 Hz, 1H), 7.14 (d, J = 8.0 Hz, 1H), 7.13 (s, 1H), 7.00 (d, J = 8.0 Hz, 1H), 6.79 (d, J = 8.0 Hz, 1H), 6.23 (s, 2H), 6.05 (s, 2H)。
1.1.5 N-[(5-溴苯并[d][1, 3]二噁英-4-基)甲基]萘并[2, 3-d][1, 3]二噁英-5-胺(6)的合成在250 mL茄型瓶中将5 (3.3 g, 8.3 mmol)溶于40 mL乙酸中, 加入二甲胺硼烷(612 mg, 10.4 mmol), 室温搅拌1 h。在经TLC (石油醚-乙酸乙酯5:1, v/v)检测反应结束后, 向反应混合液中加入200 mL水, 可见有灰白色固体析出。过滤, 滤饼经甲苯重结晶得白色粉末2.1 g, 收率63.3%。1H NMR (400 MHz, DMSO-d6) δ 7.68 (d, J = 8.4 Hz, 1H), 7.50 (s, 1H), 7.14 (s, 1H), 7.10 (dd, J = 7.6, 8.0 Hz, 1H), 7.05 (d, J = 8.4 Hz, 1H), 7.00 (d, J = 8.0 Hz, 1H), 6.44 (d, J = 7.6 Hz, 1H), 6.27 (s, 2H), 6.06 (s, 2H), 5.99 (br, 1H), 4.55 (d, J = 4.0 Hz, 2H)。
1.1.6 去甲基血根碱(7)的合成在N2保护下, 将6 (520 mg, 1.30 mmol)和Bu3SnH (783 μL, 3.90 mmol)溶于52 mL甲苯中并加热至90 ℃, 然后加入AIBN (500 mg, 2.60 mmol)并升温至110 ℃, 搅拌反应10 h。在经TLC (石油醚-乙酸乙酯5:1, v/v)监测反应结束后, 将反应液浓缩, 向浓缩后的反应液中加入10 mL石油醚, 即有沉淀析出。将沉淀过滤, 并用石油醚洗涤滤饼。此滤饼经硅胶柱色谱分离, 用石油醚-乙酸乙酯10:1 (v/v)为洗脱剂, 得亮黄色粉末218 mg, 收率52.9%。1H NMR (400 MHz, DMSO-d6) δ 9.40 (s, 1H), 8.55 (d, J = 8.9 Hz, 1H), 8.50 (s, 1H), 8.40 (d, J = 8.7 Hz, 1H), 7.99 (d, J = 8.9 Hz, 1H), 7.69 (d, J = 8.7 Hz, 1H), 7.52 (s, 1H), 6.38 (s, 2H), 6.21 (s, 2H)。ESI-MS (m/z): 318.0 [M+H]+。
1.2 5, 6-二氢-2, 3:7, 8-双亚甲二氧基-5-烷基苯并[c]菲啶的合成(8a~8f)在100 mL三口瓶中将去甲基血根碱(200 mg, 0.63 mmol)溶于不同的烷酸(20 mL)中, 在室温下搅拌, 并分批加入硼氢化钠(3.6 g, 95 mmol)。然后加热至40 ℃进行反应。在经TLC (石油醚-乙酸乙酯5:1, v/v)监测反应完全后, 用10%的NH4Cl (50 mL)淬灭反应; 将反应液用乙酸乙酯萃取两次, 每次用乙酸乙酯20 mL; 将有机相合并, 然后用饱和食盐水(50 mL)洗涤, 再经无水Na2SO4干燥后浓缩, 硅胶柱色谱分离, 用石油醚-乙酸乙酯10:1 (v/v)为洗脱剂, 得淡黄色粉末8a~8f。
1.2.1 5, 6-二氢血根碱(8a)淡黄色粉末, 产率89.3%。1H NMR (400 MHz, DMSO-d6) δ 7.77 (d, J = 8.6 Hz, 1H), 7.56 (d, J = 8.6 Hz, 1H), 7.53 (s, 1H), 7.40 (d, J = 8.2 Hz, 1H), 7.31 (s, 1H), 6.95 (d, J = 8.2 Hz, 1H), 6.15 (s, 2H), 6.09 (s, 2H), 4.13 (s, 2H), 2.52 (s, 3H)。
1.2.2 2, 3:7, 8-双亚甲二氧基-5-乙基-5, 6-二氢苯并[c]菲啶(8b)淡黄色粉末, 产率81.0%。1H NMR (400 MHz, DMSO-d6) δ 7.74 (d, J = 8.6 Hz, 1H), 7.53 (d, J = 8.6 Hz, 1H), 7.45 (s, 1H), 7.36 (d, J = 8.1 Hz, 1H), 7.29 (s, 1H), 6.91 (d, J = 8.1 Hz, 1H), 6.12 (s, 2H), 6.09 (s, 2H), 4.13 (s, 2H), 2.68 (q, J = 7.0 Hz, 2H), 1.10 (t, J = 7.0 Hz, 3H)。
1.2.3 2, 3:7, 8-双亚甲二氧基-5-(n-丙基)-5, 6-二氢苯并[c]菲啶(8c)淡黄色粉末, 产率69.7%。1H NMR (400 MHz, DMSO-d6) δ 7.74 (d, J = 8.5 Hz, 1H), 7.53 (d, J = 8.5 Hz, 1H), 7.50 (s, 1H), 7.37 (d, J = 8.1 Hz, 1H), 7.29 (s, 1H), 6.92 (d, J = 8.1 Hz, 1H), 6.12 (s, 2H), 6.08 (s, 2H), 4.13 (s, 2H), 2.60 (t, J = 7.2 Hz, 2H), 1.54~1.69 (m, 2H), 0.84 (t, J = 7.2 Hz, 3H)。
1.2.4 2, 3:7, 8-双亚甲二氧基-5-(n-丁基)-5, 6-二氢苯并[c]菲啶(8d)淡黄色粉末, 产率62.9%。1H NMR (400 MHz, DMSO-d6) δ 7.74 (d, J = 8.6 Hz, 1H), 7.53 (d, J = 8.6 Hz, 1H), 7.49 (s, 1H), 7.37 (d, J = 8.2 Hz, 1H), 7.29 (s, 1H), 6.92 (d, J = 8.2 Hz, 1H), 6.12 (s, 2H), 6.08 (s, 2H), 4.13 (s, 2H), 2.60 (t, J = 7.6 Hz, 2H), 1.57~1.49 (m, 2H), 1.33~1.22 (m, 2H), 0.84 (t, J = 7.3 Hz, 3H)。
1.2.5 2, 3:7, 8-双亚甲二氧基-5-(n-戊基)-5, 6-二氢苯并[c]菲啶(8e)淡黄色粉末, 产率48.0%。1H NMR (500 MHz, DMSO-d6) δ 7.72 (d, J = 8.4 Hz, 1H), 7.52 (d, J = 8.4 Hz, 1H), 7.49 (s, 1H), 7.35 (d, J = 8.0 Hz, 1H), 7.28 (s, 1H), 6.91 (d, J = 8.0 Hz, 1H), 6.12 (s, 2H), 6.08 (s, 2H), 4.12 (s, 2H), 2.59 (t, J = 6.9 Hz, 2H), 1.66~1.44 (m, 2H), 1.32~1.13 (m, 4H), 0.82 (t, J = 7.2 Hz, 3H)。
1.2.6 2, 3:7, 8-双亚甲二氧基-5-(n-己基)-5, 6-二氢苯并[c]菲啶(8f)淡黄色粉末, 产率31.3%。1H NMR (500 MHz, DMSO-d6) δ 7.74 (d, J = 8.6 Hz, 1H), 7.53 (d, J = 8.6 Hz, 1H), 7.49 (s, 1H), 7.36 (d, J = 8.2 Hz, 1H), 7.29 (s, 1H), 6.92 (d, J = 8.2 Hz, 1H), 6.12 (s, 2H), 6.08 (s, 2H), 4.12 (s, 2H), 2.60 (t, J = 7.4 Hz, 2H), 1.59~1.47 (m, 2H), 1.30~1.20 (m, 6H), 0.82 (t, J = 6.8 Hz, 3H)。
1.3 2, 3:7, 8-双亚甲二氧基-5-烷基苯并[c]菲啶-5-阳离子季铵盐(9a~9f)的合成在25 mL三口瓶中将烷基-去-二氢血根碱-N5-甲基衍生物粉末(0.2 mmol)溶于3 mL甲苯中, 然后加入5% NaOH水溶液1.5 mL; 将混合液搅拌均匀, 再加入DDQ (91 mg, 0.4 mmol), 继续在室温下搅拌, 并用TLC (二氯甲烷-甲醇20:1, v/v)监测反应。至反应结束后, 将反应混合液浓缩干, 将残余物溶于10 mL氯仿中, 然后滴加浓盐酸(1 mL); 将析出的红色沉淀过滤, 收集滤饼, 用二氧六环进行重结晶后得化合物9a~9f。
1.3.1 血根碱(9a)红色粉末, 产率90.0%。1H NMR (500 MHz, DMSO-d6) δ 10.13 (s, 1H), 8.78 (d, J = 9.0 Hz, 1H), 8.64 (d, J = 8.7 Hz, 1H), 8.30 (d, J = 9.0 Hz, 1H), 8.27 (s, 1H), 8.12 (d, J = 8.7 Hz, 1H), 7.77 (s, 1H), 6.60 (br s, 2H), 6.34 (br s, 2H), 4.91 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 150.0, 148.8 (×2), 147.6, 146.3, 132.2, 131.5, 131.4, 127.2, 125.7, 120.3, 120.0, 118.9, 117.4, 109.5, 105.8, 105.0, 104.3, 102.9, 52.2; ESI-MS (m/z) 332.1 [M-Cl]+。
1.3.2 2, 3:7, 8-双亚甲二氧基-5-乙基苯并[c]菲啶(9b)红色粉末, 产率93.0%。1H NMR (400 MHz, DMSO-d6) δ 10.17 (s, 1H), 8.77 (d, J = 8.8 Hz, 1H), 8.62 (d, J = 8.8 Hz, 1H), 8.29 (d, J = 8.8 Hz, 1H), 8.11 (d, J = 8.8 Hz, 1H), 8.00 (s, 1H), 7.76 (s, 1H), 6.60 (s, 2H), 6.33 (s, 2H), 5.29 (q, J = 6.7 Hz, 2H), 1.66 (t, J = 6.7 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 149.7, 149.5, 149.3, 148.1, 147.0, 132.5, 131.7, 131.1, 127.8, 126.5, 120.6, 120.5, 119.4, 117.8, 110.4, 106.3, 105.4, 103.6, 103.3, 58.1, 16.5; ESI-MS (m/z) 346.1 [M-Cl]+。
1.3.3 2, 3:7, 8-双亚甲二氧基-5-(n-丙基)苯并[c]菲啶(9c)红色粉末, 产率85.3%。1H NMR (400 MHz, DMSO-d6) δ 10.21 (s, 1H), 8.77 (d, J = 8.8 Hz, 1H), 8.62 (d, J = 8.8 Hz, 1H), 8.29 (d, J = 8.8 Hz, 1H), 8.11 (d, J = 8.8 Hz, 1H), 8.02 (s, 1H), 7.76 (s, 1H), 6.60 (s, 2H), 6.33 (s, 2H), 5.29 (t, J = 7.2 Hz, 2H), 2.04~1.90 (m, 2H), 0.77 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 149.9, 149.6, 149.4, 148.2, 147.2, 132.4, 131.8, 131.0, 127.6, 126.4, 120.70, 120.65, 119.4, 117.8, 110.3, 106.2, 105.4, 103.4, 103.3, 66.8, 24.5, 10.6; ESI-MS (m/z) 360.1 [M-Cl]+。
1.3.4 2, 3:7, 8-双亚甲二氧基-5-(n-丁基)苯并[c]菲啶(9d)红色粉末, 产率74.7%。1H NMR (400 MHz, DMSO-d6) δ 10.23 (s, 1H), 8.77 (d, J = 8.8 Hz, 1H), 8.62 (d, J = 8.8 Hz, 1H), 8.29 (d, J = 8.8 Hz, 1H), 8.11 (d, J = 8.8 Hz, 1H), 8.02 (s, 1H), 7.76 (s, 1H), 6.60 (s, 2H), 6.33 (s, 2H), 5.29 (t, J = 7.2 Hz, 2H), 2.01~1.87 (m, 2H), 1.15~1.02 (m, 2H), 0.78 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 149.9, 149.5, 149.4, 148.2, 147.2, 132.4, 131.8, 131.0, 127.6, 126.4, 120.7, 120.6, 119.4, 117.8, 110.3, 106.3, 105.4, 103.5, 103.3, 62.3, 32.9, 19.3, 13.6; ESI-MS (m/z) 374.1 [M-Cl]+。
1.3.5 2, 3:7, 8-双亚甲二氧基-5-(n-戊基)苯并[c]菲啶(9e)红色粉末, 产率68.2%。1H NMR (400 MHz, DMSO-d6) δ 10.23 (s, 1H), 8.76 (d, J = 8.9 Hz, 1H), 8.62 (d, J = 8.9 Hz, 1H), 8.28 (d, J = 8.9 Hz, 1H), 8.11 (d, J = 8.9 Hz, 1H), 8.01 (s, 1H), 7.75 (s, 1H), 6.60 (s, 2H), 6.33 (s, 2H), 5.27 (t, J = 7.2 Hz, 2H), 2.02~1.90 (m, 2H), 1.25~1.14 (m, 2H), 1.14~1.04 (m, 2H), 0.74 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 149.9, 149.5, 149.3, 148.2, 147.2, 132.4, 131.7, 130.3, 127.6, 126.4, 120.6, 119.4, 117.8, 110.3, 106.3, 105.4, 103.4, 103.3, 102.1, 62.5, 30.6, 28.0, 21.8, 14.1; ESI- MS (m/z) 388.2 [M-Cl]+。
1.3.6 2, 3:7, 8-双亚甲二氧基-5-(n-己基)苯并[c]菲啶(9f)红色粉末, 产率72.4%。1H NMR (400 MHz, DMSO-d6) δ 10.23 (s, 1H), 8.76 (d, J = 8.7 Hz, 1H), 8.62 (d, J = 8.7 Hz, 1H), 8.28 (d, J = 8.7 Hz, 1H), 8.11 (d, J = 8.7 Hz, 1H), 8.01 (s, 1H), 7.75 (s, 1H), 6.60 (s, 2H), 6.33 (s, 2H), 5.28 (t, J = 7.2 Hz, 2H), 1.99~1.88 (m, 2H), 1.17~1.04 (m, 6H), 0.73 (t, J = 6.3 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 149.9, 149.5, 149.4, 148.2, 147.2, 132.4, 131.7, 131.0, 127.6, 126.4, 120.7, 119.4, 117.8, 110.3, 106.2, 105.4, 103.4, 103.3, 102.1, 62.5, 30.8, 30.7, 25.4, 22.2, 14.1; ESI-MS (m/z) 402.2 [M-Cl]+。
2 肿瘤细胞株生长抑制实验将待测化合物用100 μL DMSO溶解并用含10%血清的1640培养基将待测化合物母液稀释至工作液浓度。将人相应的肿瘤细胞制成细胞悬液, 按一定浓度加入96孔培养板(100 μL/孔), 置于37 ℃、5% CO2的孵箱培养24 h。加入待测化合物的工作液, 其终浓度为0.1、1、10、100 μmol·L-1 (每个浓度4个复孔)。作用72 h后弃去培养液, 每孔加入100 μL含0.5 mg·mL-1 MTT的1640培养基(10%血清), 置于37 ℃、5% CO2的孵箱培养4 h后弃液, 每孔加入DMSO 150 μL, 常温下振摇10 min, 使蓝色结晶完全溶解, 以检测波长570 nm、参考波长655 nm于Bio- Rad 450型酶标仪测定每孔吸光度。按下列公式计算待测品的抑制率:
| $ \begin{array}{l} {\rm{待测化合物生长抑制率}}\left( \% \right) = \frac{{阴性对照吸光度 - 待测化合物吸光度}}{{阴性对照吸光度 - 本底吸光}} \times 100\% \\ \end{array} $ |
IC50值按化合物各浓度相应的抑制率和剂量的对数做图并计算。本文以上市药物5-Fu为阳性对照进行活性评价, 结果如表 1所示。
| Table 1 Cancer cells growth inhibitory activity of the target compounds |
| [1] | Schmeller T, Latz-Brüning B, Wink M. Biochemical activities of berberine, palmatine and sanguinarine mediating chemical defence against microorganisms and herbivores[J]. Phytochemistry, 1997, 44: 257–266. DOI:10.1016/S0031-9422(96)00545-6 |
| [2] | Hu Z, Xu Y, Feng S, et al. Studies on the active principles of the fruits of Macleaya cordata (Willd.) R. Br.[J]. Acta Pharm Sin (药学学报), 1979, 14: 535–540. |
| [3] | Ye F, Feng F, Liu W. Alkaloids from Macleaya cordata[J]. China J Chin Mater Med (中国中药杂志), 2009, 34: 1683–1686. |
| [4] | Qin H, Wang P, Li Z, et al. The establishment of the control substance and 1H NMR nuclear magnetic resonance fingerprint of Macleaya microcarpa (Maxim.) Fedde[J]. Chin J Anal Chem (分析化学), 2004, 32: 1165–1170. |
| [5] | Oechslin SM, K nig GM, Oechslin-Merkel K, et al. An NMR study of four benzophenanthridine alkaloids[J]. J Nat Prod, 1991, 54: 519–524. DOI:10.1021/np50074a026 |
| [6] | Zhou JY, Chen BZ, Tong XJ, et al. Chemical study on Chelidonium majus alkaloids[J]. Chin Tradit Herb Drugs (中草药), 1989, 20: 2–4. |
| [7] | Zuo JL, Bai L, Song XX, et al. Simultaneous determination of sanguinarine, berberine and chelerythrine in Chelidonium majus by RP-HPLC[J]. Chin J Pharm Anal (药物分析杂志), 2008, 28: 903–905. |
| [8] | Caballero-George C, Vanderheyden PML, Apers S, et al. Inhibitory activity on binding of specific ligands to the human angiotensin Ⅱ AT1 and endothelin 1 ETA receptors:bioactive benzo[J]. Planta Med, 2002, 68: 770–775. DOI:10.1055/s-2002-34406 |
| [9] | Dai B, Zhang MJ, Tian SJ, et al. Research progress on application and pharmacological activity of sanguinarine[J]. J Tradit Chin Vet Med (中兽医医药杂志), 2015(4): 73–75. |
| [10] | Caballero-George C, Vanderheyden PML, Solis PN, et al. In vitro effect of sanguinarine alkaloid on binding of[J]. Eur J Pharmcol, 2003, 458: 257–262. DOI:10.1016/S0014-2999(02)02819-4 |
| [11] | Ahsan H, Reagan-Shaw S, Breur J, et al. Sanguinarine induces apoptosis of human pancreatic carcinoma AsPC-1 and BxPC-3 cells via modulations in Bcl-2 family proteins[J]. Cancer Lett, 2007, 249: 198–208. DOI:10.1016/j.canlet.2006.08.018 |
| [12] | Ping G, Wang Y, Shen L, et al. Highly efficient complexation of sanguinarine alkaloid by carboxylatopillararene:pKa shift, increased solubility and enhanced antibacterial activity[J]. Chem Commun, 2017, 53: 7381–7384. DOI:10.1039/C7CC02799K |
| [13] | Janovská M, Kubala M, Šimánek V, et al. Fluorescence of sanguinarine:fundamental characteristics and analysis of interconversion between various forms[J]. Anal Bioanal Chem, 2009, 395: 235–240. DOI:10.1007/s00216-009-2903-9 |
| [14] | Nakanishi T, Suzuki M, Mashiba A, et al. Synthesis of NK109, an anticancer benzo[c]phenanthridine alkaloid[J]. J Org Chem, 1998, 63: 4235–4239. DOI:10.1021/jo9718758 |
| [15] | Maestri G, Larraufie MH, Derat Ĕ, et al. Expeditious synthesis of phenanthridines from benzylamines via dual palladium catalysis[J]. Org Lett, 2010, 12: 5692–5695. DOI:10.1021/ol102509n |
| [16] | Ishii H, Ishikawa T, Watanabe T, et al. Conversion of the naturally occurring amide alkaloids into O5 benzo[c]phenanthridinium alkaloids. A new synthetic sequence to antitumor benzo[c]phenanthridine alkaloids[J]. J Chem Soc, Perkin Trans I, 1984: 2283–2289. DOI:10.1039/p19840002283 |
| [17] | Harayama T. Synthesis of benzo[c]phenanthridine alkaloids using a palladium-catalyzed aryl-aryl coupling reaction[J]. Heterocycles, 2005, 65: 697–713. DOI:10.3987/REV-04-594 |
| [18] | De S, Mishra S, Kakde BN, et al. Expeditious approach to pyrrolophenanthridones, phenanthridines, and benzo[c] phenanthridines via organocatalytic direct biaryl-coupling promoted by potassium tert-butoxide[J]. J Org Chem, 2013, 78: 7823–7844. DOI:10.1021/jo400890k |
| [19] | Ishii H, Ishikawa T, Ichikawa YI, et al. Studies on the chemical constituents of Rutaceous plantss. LV. The development of a versatile method for the synthesis of antitumor active benzo[c] phenanthridine alkaloids. (5). A new method for quaternization of the benzo[c]phenanthridine nucleu[J]. Chem Pharm Bull, 1984, 32: 2984–2994. DOI:10.1248/cpb.32.2984 |
| [20] | Nakanishi T, Suzuki M, Saimoto A, et al. Structural considerations of NK109, an antitumor benzo[c]phenanthridine alkaloid[J]. J Nat Prod, 1999, 62: 864–867. DOI:10.1021/np990005d |
| [21] | Guo J, Dong W, Liu W, et al. Synthesis and cytotoxicity of 3-aryl acrylic amide derivatives of the simplified saframycin-ecteinascidin skeleton prepared from L-dopa[J]. Eur J Med Chem, 2013, 62: 670–676. DOI:10.1016/j.ejmech.2013.01.033 |