 2018, Vol. 53
2018, Vol. 53

新药发现与研究实例简析
新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和疗效评价等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
根据PDE5酶底物的结构特征, 选定具有相似结构的化合物作为苗头分子, 对结构的“东西南北中”作广泛的变换和构效关系研究, 优化活性强度、选择性和成药性等。西地那非的首创意义在于临床研究中“偶然发现”可以临时克服男性勃起障碍, 虽然原定研发心血管药物的目标没有达到, 却划时代地发明了第一个口服治疗男性勃起障碍的药物, 在一定程度上改善了人们的生活质量, 其首创性意义重大。失之东隅, 收之桑榆, 是因为有相同的机制和作用靶标, 彰显了偶遇的必然性。
(编者按)
一氧化氮(NO)是个化学不稳定的无机分子, 却广泛存在于人体细胞中, 是由精氨酸与O2在一氧化氮合酶催化下生成的(也可由硝酸盐转变成NO)。NO作为气体分子可迅速穿越细胞膜, 在细胞间传递, 半衰期短(多数内源性介质的共同特征, 履行完生理功能后即行消失)。NO在体内具有多种生理功能, 其中一个重要的作用是在平滑肌细胞内激活鸟苷酸环化酶, 该酶将三磷酸鸟苷(GTP)转变为环鸟苷酸(cGMP), 因而NO可提高胞内cGMP水平, 后者又启动一系列级联反应, 最终降低了胞内钙, 引起平滑肌细胞松弛, 使血管舒张, 从而降低了外周血管阻力, 所以NO可改善心肌缺血状态。然而长时间使用硝酸盐会迅速耐受而失效。辉瑞的科学家希望找到其他可调控NO水平的途径。
cGMP是体内无处不在的第二信使, 在细胞内水平受环核苷酸酶(催化合成)和磷酸二酯酶(催化降解)调控, 促进NO释放或抑制磷酸二酯酶(PDE)可提高cGMP水平。PDE水解cGMP成无活性的GMP。
1.2 磷酸二酯酶PDEs是一个多基因的大家族, 它包括11类亚型共30余种, 在细胞与亚细胞区域作不同分布。PDEs家族有相似的结构, 都包含调控和催化两个功能区域。各型PDEs催化域的氨基酸序列相似75%以上。显示出家族成员间的同源性。PDEs具有不同的底物特异性: PDE4、7和8水解cAMP, 而PDE5、6和9选择性水解cGMP。PDE1和2既能水解cAMP, 也能水解cGMP。
1.3 PDE5活性中心的特征在研发西地那非时, PDE5的三维结构尚未解析, 后来经X-射线衍射分析揭示了PDE5的结构。PDE5的催化位点处于C端螺旋束的中心, 结合底物的腔穴大约10 Å深, 口窄内宽, 体积为330 Å3, 由4个亚基组成:金属结合位点、母核结合腔、疏水腔和顶盖区域。金属结合位点含有锌离子和镁离子。锌离子结合于氨基酸残基的His 617 (键长2.1 Å)、Asp 654 (2.1 Å)、Asp 764 (2.2 Å)、His 653 (2.1 Å)和两个水分子(W1 2.2 Å, W2 2.8 Å); 镁离子与Asp 654 (2.0 Å)和5个水分子形成配位键(2.0, 2.0, 2.1, 2.0, 2.6 Å)。母核结合腔容纳cGMP的鸟嘌呤; 疏水腔由Phe 786、Ala 783、Leu 804和Val 782组成, 容纳核糖部分; 顶盖区域在开口处, 由Tyr 664、Met 816、Ala 823和Gly 819构成。这些腔穴都在PDE5抑制剂的结合中体现了出来。
1.4 男性勃起障碍生理学研究提示, 男性的勃起机制是当男性受到性刺激, 阴茎海绵体细胞内产生NO, NO刺激cGMP的生成, 后者导致勃起组织的螺旋动脉内膜的垫平滑肌松弛, 舒张的平滑肌增加了阴茎海绵组织的血流量, 导致勃起。勃起障碍常常与冠状动脉病平行发生, 是因为生化机制相同的缘故, 也决定了辉瑞意外发现西地那非是必然的。
PDE亚型的组织分布具有特异性, PDE5主要分布在阴茎海绵体平滑肌和血管平滑肌内, 所以PDE5是治疗勃起障碍的药物靶标。不过, 当初辉瑞研发PDE5的抑制剂是以心血管疾病为目标, 并未意识到治疗勃起障碍。
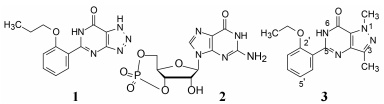
2 苗头化合物扎普司特为了研发以PDE5为靶标的抗肺动脉高压药物, 研究者考虑到扎普司特(1, zaprinast)与cGMP (2)的结构相似性而作为起始物。扎普司特是M & Baker公司(后并入安万特公司)研制上市的抗过敏药, 具有解除平滑肌痉挛抗哮喘作用。1991年Haynes等发现扎普司特可降低大鼠因肺泡缺氧而血管收缩得到缓解, 还发现可舒张肺动脉血管, 这些可能与抑制支气管平滑肌的PDE5有关。然而1对多种PDE亚型有抑制作用, 而抑制PDE5的活性强度和选择性都不高。辉瑞设定的研究目标是通过提高对PDE5的选择性抑制作用, 研制新结构类型的降低肺动脉高压等心血管药物。
为了评价化合物的活性和选择性, 用自家兔血小板中分离的PDE5作为筛选活性的模型; 用大鼠肝的PDE1和家兔血小板的PDE3等模型评价化合物的选择性作用。辉瑞的药化合成五人小组在三年中合成并评价了1 600个化合物, 通过分析构效关系, 逐步优化了化合物的活性、选择性、物化和药代性质等。以下是由苗头化合物扎普司特开始经骨架迁越和药效团的探索, 最终研发出西地那非的过程, 所列的化合物是各个阶段有代表性的化合物。
通过对化合物1的三唑并嘧啶酮母核一系列变换, 发现三唑环变换成甲基吡唑化合物3, 对PDE5的活性显著提高, 3对PDE5的选择性10倍于PDE1。表 1列出了1和3抑制PDEs活性数据。因而将化合物3作为先导化合物进行优化(Terrett NK, Bell AS, Brown D, et al. Sildenafil (Viagra(TM)), a potent and selective inhibitor of type 5 cGMP phosphodiesterase with utility for the treatment of male erectile dysfunction. Bioorg Med Chem Lett, 1996, 6: 1819-1824)。
| Table 1 Comparison of inhibitions on PDEs between zaprinast (1) and compound 3 |
化合物3的骨架是吡唑并嘧啶酮, 经分子模拟对3与鸟苷的母核作结构比较, 表明分子尺寸、形状和偶极矩具有相似性。
|
分析化合物3的母核结构, 3位甲基的处境相当于鸟苷酸的核糖, 因而做出的科学假设是, 甲基履行了核糖片段与受体发生疏水性结合的功能(核糖整体虽是亲水性的, 但糖环面向受体的一侧是疏水性的, 许多酪氨酸激酶抑制剂的设计也如是)。甲基的体积小于戊糖环, 未能充满该疏水腔穴, 故而将3位甲基加大为正丙基, 得到化合物4的活性和选择性都有显著提高。5位存在有取代的苯环是必要的, 它所处的空间相当于cGMP磷酸结合的位置。
N1的甲基对PDE5的选择性很重要, 去除甲基则降低了对PDE5的活性。例如化合物5对PDE5的抑制作用弱于4(表 2)。

|
| Table 2 Inhibitory activities of compounds 4 and 5 on PDE enzymes |
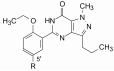
为了优化5位苯环上的取代基, 保持母核上N1-甲基和C3-正丙基不变, 变换苯环上2'的乙氧基, 用羟基、硝基和甲磺酰胺基置换, 以及用环丙甲氧基取代, 都使活性下降, 表明乙氧基是重要的基团。表 3列出了有代表性的化合物6~10对不同亚型PDE的活性和选择性。
| Table 3 Inhibitions of compounds 6-10 on PDE1, 3 and 5. NT = Not test |


为了证明2'位乙氧基对活性的优势, 分子模拟和X-射线晶体学研究显示, 乙氧基的氧孤电子对与N6的氢原子形成分子内氢键, 提高了苯环与母核的共面性, 并且连接尺寸大的疏水性基团不利于活性。
3.3 5'位的优化——候选物西地那非的确定至此化合物4成为先导物优化的最佳结果, 但尚未探索苯环5'位置的取代基对活性的影响, 加之由于化合物4的脂溶性高于水溶性10 000倍, 水溶性较低, 不利于口服吸收, 需要在5'位置加入极性基团以利于成药性质。
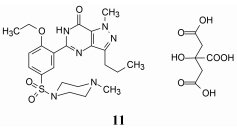
在苯环上变换或引入基团有三方面的考虑:一是由于这个位置相当于cGMP的磷酯基, 引入模拟磷酯的基团可提高与PDE5酶的互补性结合; 二是加入极性或成盐基团以有利于水溶性; 再一是在研发扎普司特的构效关系时发现, 5'位存在有磺酰基或磺酰胺基可提高抗过敏活性, 提示这个位置可容纳新的基团(Wilson SR, Wilson RB, Shoemaker AL, et al. Antiallergic 8-aza-purines 3. Structural characterization of 2-(2-propoxyphenyl)-8-azahypoxanthine, 2-(2-propoxy- 5-(propylsulfonyl) phenyl)-8-azahypoxanthine, and 2-(2- propoxy-5-(N-methyl-N-isopropylsulfamoyl)phenyl)- 8-azahypoxanthine. J Am Chem Soc, 1982, 104: 259-264)。为此, 在5'位连接(取代的)哌嗪或哌啶基, 得到代表性的化合物11~14, 其结构、活性和分布系数列于表 4。化合物11~14对PDE5的抑制活性和选择性作用明显高于5'位没有取代的化合物4, 而且溶解性提高了数十倍, 提示设计思路的正确性。其中化合物11的体内活性、选择性和药代性质均优于其他化合物, 遂制成枸橼酸盐, 定名为西地那非(sildenafil)进入临床研究(Ghofrani HA, Osterloh IH, Grimminger F. Sildenafil: from angina to erectile dys- function to pulmonary hypertension and beyond. Nat Rev Drug Discov, 2006, 5: 689-702)。
| Table 4 Effects of 5'-substituent on the activity and solubility |




在结构优化、候选物的遴选以及临床前的研究, 辉瑞公司的目标是针对PDE5靶标研发治疗冠心病和肺动脉高压药物, 枸橼酸西地那非(11)在经过健康志愿者Ⅰ期试验后, 进入Ⅱ期试验阶段, 对有限数量的心绞痛患者进行实验治疗观察, 结果对心绞痛患者没有达到预期的效果, 因而终止了对治疗心血管病的临床研究。
4.2 偶遇的必然性—西地那非上市以Osterloh为首的临床试验人员在分析归纳西地那非的临床效果时, 意外地发现了一些男性受试者服药后生殖器出现勃起现象, 勃起程度与服用剂量呈相关性, 高剂量实验组中近90%的人都有勃起现象。这个发现促使辉瑞公司改变了该项目的适应症研究, 在1994年对12个有勃起障碍的患者进行了Ⅱ期临床研究, 结果10名患者的勃起有显著增强。
|
枸橼酸西地那非的口服生物利用度F = 40%, 血浆达峰时间为1 h, 半衰期t1/2 = 2 h, 肝脏中主要经CYP3A4代谢, 发生N-去甲基化, 去甲基代谢产物仍具有抑制PDE5作用, 但强度只是原药的50%。
经过Ⅲ期临床研究, 证实西地那非有确实的促进男性勃起的功能, 于1998年FDA批准作为促进勃起的药物上市。西地那非的发现, 比当初旨在治疗心血管病的意义更大, 因为它是首创性地通过口服途径、特异性地抑制PDE5提高了勃起能力, 不同于既往的药物经过性激素介导或神经刺激的方式, 这在一定的程度上改善了生活质量, 并由此开创了以PDE5为靶标的治疗勃起障碍的药物新领域。
4.3 西地那非的不良反应西地那非是以舒张血管为治疗心血管病而研制的, 在临床阶段改变了适应症成为治疗勃起功能障碍的药物。因而西地那非存留的引起低血压成为它的不良反应:健康男性志愿者单剂口服西地那非, 导致卧位血压下降, 服药后1~2 h血压下降最明显, 8 h则与安慰剂组无差别。若同时服用硝酸酯类药物, 降压作用更大, 是因为提供了NO的来源。另一不良反应是对视觉的影响, 即出现一过性蓝/绿颜色辨别异常, 异常程度与剂量相关, 这是因为西地那非具有抑制PDE6 (IC50 37 nmol·L-1)的作用。PDE6参与视网膜中光传导的生理过程。虽然西地那非对PDE6抑制作用弱于PDE5大约10倍, 而且是一过性的, 但勃起的同时有蓝视障碍仍是不适宜的。
4.4 西地那非与PDE5的结合模式在研发西地那非时, PDE5的三维结构尚未解析, 优化结构是靠传统药物化学方法进行的。后续研究的西地那非与PDE5复合物晶体结构的结合特征表明, 结构骨架和周边的侧链和基团与活性中心呈互补性结合。图 1是西地那非与PDE5复合物的晶体结构立体图。结合的主要特征如下:①母核吡唑并嘧啶酮与cGMP的鸟嘌呤环相似, 结合于Gln 817、Phe 820、Val 782和Tyr 612构成的疏水腔中, 吡唑环与其中的Phe 820发生π-π叠合作用。②吡唑环的N2与水分子形成氢键, 该水分子又同时与Tyr 612和另一个与锌离子配位的水分子形成氢键网络。③嘧啶酮环的C=O和N-H分别与Gln 817形成双齿型氢键, 前者是氢键接受体, 后者是氢键给体。④由Gln 817-Gln 775、Gln 775-Ala 767和Gln 775-Trp 853形成的氢键网络将5位苯环上的两个侧链的走向和氢键的形成起到引导和稳固作用。⑤甲基哌嗪处于活性中心的开口的顶盖处, 磺酰基没有发生氢键结合(Sung BJ, Hwang KY, Jeon YH, et al. Structure of the catalytic domain of human phosphodiesterase 5 with bound drug molecules. Nature, 2003, 425: 98-102)。

|
Figure 1 Stereoviews of crystallography of complex sildenafil with PDE5 |
拜耳公司快速跟踪研制的伐地那非(15, vardenafil), 采用了骨架迁越和保持药效团的策略, 新骨架是用咪唑并三嗪酮替换西地那非的吡唑并嘧啶酮环, 是将吡唑环的N1与并环处的C交换, 形成了新的环系, 赋予化合物以结构新颖性。其实这两个骨架都是cGMP的鸟嘌呤环电子等排体。伐地那非骨架上连接的基团模拟了西地那非的药效团。研制过程也证实了那些药效团特征是呈现活性的必要基团。例如5'无取代基的化合物16抑制PDE5的IC50 = 5 nmol·L-1, 伐地那非的IC50 = 1.2 nmol·L-1 (Hanning H, Niewöhner U, Schenke T, et al. Imidazo[5, 1-f][1, 2, 4] triazin-4(3H)-ones: a new class of potent PDE5 inhibitors. Bioorg Med Chem Lett, 2002, 12: 865-868)。伐地那非的口服生物利用度F = 15%, 半衰期t1/2 = 4~5 h。2003年FDA批准上市。
5.2 乌地那非乌地那非(17, udenafil)是韩国东亚药业研发的治疗勃起障碍药物, 作用靶标也是PDE5。乌地那非结构骨架与西地那非相同, 药效团特征也大同小异, 是个典型的跟进型药物。乌地那非抑制PDE5的IC50 = 5.2 nmol·L-1, 口服后迅速起效, tmax = 1.0~1.5 h, 半衰期t1/2 = 10~12 h (Kim BH, Lim HS, Chung JY, et al. Safety, tolerability and pharmacokinetics of udenafil, a novel PDE-5 inhibitor, in healthy young Korean subjects. Brit J Clin Pharmacol, 2008, 65: 848-854)。
5.3 他达那非LillyIcos公司研发的他达那非(18, tadanafil)是第三个上市的强效的PDE5抑制剂, IC50 = 1~5 nmol·L-1, 其结构类型与上述的“那非”药物不同。主要用于男性勃起障碍, 也用于治疗肺动脉高压和尿道功能障碍。用药剂量10 mg, 起效时间内15~30 min, 半衰期t1/2 = 44~112 h。

|
阿伐那非(19, avanafil)是田边制药研发的PDE5抑制剂, 美国FDA于2012年批准上市, 体外抑制PDE5活性IC50 = 1 nmol·L-1, 口服15~30 min即发生勃起(Limin M, Johnsen N, Hellstrom WJ. Avanafil, a new rapid-onset phosphodiesterase 5 inhibitor for the treatment of erectile dysfunction. Expert Opin Investig Drugs, 2010, 19: 1427-1437)。