 2018, Vol. 53
2018, Vol. 53



2. 河北大学 药物化学与分子诊断教育部重点实验室, 河北 保定 071002;
3. 北京大学药学院天然药物及仿生药物国家重点实验室药物设计中心, 北京 100191
2. Key Laboratory of Medicinal Chemistry and Molecular Diagnosis of Ministry of Education, Hebei University, Baoding 071002, China;
3. Drug Design Center, State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, Beijing 100191, China
真菌是一类分布十分广泛的真核细胞生物, 它们与人类关系密切, 其中许多真菌对人类是有益的, 然而还有些真菌可侵犯动植物及人类, 引起各种真菌病[1, 2]。虽然人们对于一些结构新颖、活性突出的抗真菌药物需求量巨大, 但是抗真菌药物的发展依然十分缓慢[3, 4]。甾醇14α-去甲基化酶(sterol 14α- demethylase, CYP51)是生物甾醇合成过程中的一个关键酶, 为细胞色素P450超家族的主要成员之一[5]。其主要功能是催化羊毛甾醇14α-位的甲基离去[6]。一旦此酶缺乏或其功能受到抑制, 生物体就无法合成甾醇, 细胞膜无法合成, 最终细胞因失去细胞膜而死亡[7]。自然界中, CYP51广泛存在于植物、真菌、哺乳动物体内, 是氮唑类抗真菌药物的靶标酶, 在药物的设计和研发中占有重要地位[8, 9]。
噻二唑类化合物是由包括氢键结合域、硫原子和双电子供体氮系统组成的五元杂环类化合物, 表现出广泛的生物活性[10]。噻二唑类在自然界中存在以下4种异构体形式:分别是1, 2, 3-噻二唑、1, 2, 5-噻二唑、1, 2, 4-噻二唑和1, 3, 4-噻二唑[11, 12]。其中, 1, 3, 4-噻二唑类的化合物对各种病原体显示出广谱的活性[13]。这些化合物通过竞争性抑制真菌甾醇生物合成中的关键酶CYP51发挥作用[14]。CYP51的选择性抑制会引起麦角甾醇的消耗以及羊毛甾醇和其他14-甲基甾醇的积累, 导致真菌细胞的生长抑制[15, 16]。
鉴于1, 3, 4-噻二唑和硫色满酮杂环类化合物均具有良好生物活性[17], 根据药物设计的生物活性基团拼接原理[18-20], 设计了一系列的(E)-3-{[(1, 3, 4-噻二唑-2-基)氨基]亚甲基}-硫色满-4-酮类衍生物, 采用微量稀释法评价其抗真菌活性, 得出初步构效关系, 并通过分子对接探讨该类化合物与白色念珠菌的甾醇14α-去甲基化酶的结合方式, 为进一步结构改造提供依据[21, 22]。
结果与讨论 1 化合物的合成及结构鉴定本文以5a为例, 由化合物3、4合成化合物5a的过程中(合成路线1), 考察了不同催化剂、溶剂对反应速率的影响。
以乙醇作溶剂, 考察了三乙胺、氢氧化钠、醋酸、盐酸等不同催化剂对产率的影响, 结果表明, 盐酸作催化剂时产率最高为83%。接着在盐酸作催化剂的条件下, 考察了乙醇、水、二氯甲烷等溶剂对反应产率的影响, 结果当乙醇作溶剂时, 产率最高。合成的最佳反应条件确定为:乙醇为溶剂, 盐酸为催化剂。

|
Scheme1 Synthesis of (E)-3-(((1, 3, 4-thiadiazol-2-yl)amino)methylene)-thiochroman-4-ones |
目标化合物经过HR-MS、1H NMR、13C NMR等方法进行了表征, 理化数据见表 1、2。1D-noesy谱图中-S-CH2- (δH 3.81)和-C=CH- (δH 7.83) (化合物5h)之间存在NOE效应, 说明化合物的双键为E构型。
| 表 1 Structure and yield of synthesized compounds 5a-5u |


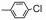
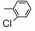
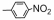
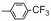


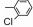
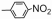
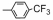

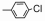


| Table 2 The spectral data of compounds 5a-5u |
以辣椒炭疽病菌(Colletotrichum capsici, C.c.)、小麦纹枯病菌(Rhizoctonia cerealis, R.c.)、苹果树腐烂病(Valsa mali, V.m.)、花生冠腐病菌(Aspergillus niger, A.n.)为供试菌。对合成的目标化合物5a~5u的抑菌活性进行初步筛选, 结果见表 3。结果表明化合物5n、5o对R.c.抗菌活性优于阳性对照多菌灵(carbendazim, CBD), 最小抑菌浓度均为16 μg·mL-1。化合物5j对C.c.、R.c.、A.n.的抗菌活性也优于阳性对照多菌灵; 5p对R.c.、V.m.、A.n.抗菌活性与阳性对照多菌灵相当。化合物5a、5d、5r和5t表现出一定的抗菌活性, 但是, 所有化合物的活性均显著低于两性霉素B (amphotericin B, AmB)。
| Table 3 Minimum inhibitory concentrations (MIC/μg·mL-1) of target compounds. "-" indicated no inhibitory activities; Colletotrichum capsici (C.c.), Valsa mali (V.m.), Aspergillus niger (A.n.), Rhizoctonia cerealis (R.c.) |
以新生隐球菌(Cryptococcus neoformans, C.n.)、白色念珠菌(Canidia albicans, C.a.)、烟曲霉(Asper gillus funigatus, A.f.)、絮状表皮癣菌(Epidermophyton floccosum, E.f.)、总状毛霉菌(Mucor racemosus, M.r.)和须癣毛癣菌(Trichophyton mentagrophytes, T.m.)为供试对象, 对合成的目标化合物5a~5u进行抑菌活性测试, 结果见表 4, 总体来看, 化合物对C.n.、C.a.和A.f.的抗菌活性要优于M.r.、T.m.和E.f.。化合物5h抗菌活性最强, 对C.a.最小抑菌浓度为8 μg·mL-1, 对C.n.、A.f.、E.f.最小抑菌浓度为16 μg·mL-1; 其次是5i, 对C.a.、A.f.和M.r.的最小抑菌浓度均为16 μg·mL-1; 化合物5o对C.a.、A.f.最小抑菌浓度均为16 μg·mL-1; 化合物5a对A.f.的最小抑菌浓度为16 μg·mL-1, 对C.a.最小抑菌浓度为32 μg·mL-1, 均优于阳性对照氟康唑。其余化合物对被试菌株也表现出一定的抗菌活性, 但所有化合物的抗真菌活性均显著低于两性霉素B。
| Table 4 Minimum inhibitory concentrations (MIC/μg·mL-1) and C-score of target compounds. "-" indicated no inhibitory activities; Cryptococcus neoformans (C.n.), Canidia albicans (C.a.), Aspergillus funigatus (A.f.), Mucor racemosus (M.r.), Epidermophyton floccosum (E.f.) and Trichophyton mentagrophytes (T.m.) |
R2为烷烃时的抗菌活性优于芳香烃, 可能的原因为芳香烃空间位阻大, 无法进入活性口袋, 从而使化合物无法与作用靶点进行结合, 分子对接结果也证明了这点。另外, 当硫色满酮的苯环上带有吸电子取代基时(如氟、氯)、1, 3, 4-噻二唑的2位为供电子取代基时(如甲基或乙基), 化合物表现出较好的抗菌活性, 这对进一步设计工作具有指导意义。
4 分子对接研究目标化合物5a~5u、阳性对照氟康唑分别与白色念珠菌的甾醇14α-去甲基化酶(ID: 5TZ1)的对接结果见表 4。对接结果表明:化合物的对接打分值(C-score)与其抗真菌活性大体相一致。
图 1、图 2和图 3为半柔性分子对接研究得到的5h、5c和氟康唑分别与CYP51活性位点的相互作用模式。从图中可以看出, 当R2位为烷烃时(图 1), 化合物可以与CYP51的血红素铁原子形成配位结合, 而当R2位为芳香烃时(图 2), 可能是因为苯环的空间位阻过大, 导致化合物无法与靶点的血红素铁原子形成配位结合。

|
Figure 1 Predicted binding mode of 5h docked into the binding site of CYP51 14α-sterol demethylase of Candida albicans |

|
Figure 2 Predicted binding mode of 5c docked into the binding site of CYP51 14α-sterol demethylase of Candida albicans |

|
Figure 3 Predicted binding mode of fluconazole docked into the binding site of CYP51 14α-sterol demethylase of Candida albicans |
同时, 分子对接研究表明, 化合物与CYP51主要通过氢键、疏水和范德华力相互作用。另外, 噻二唑环上的氮原子能与CYP51上的血红素铁原子形成配位结合, 说明化合物中噻二唑环为抗真菌活性的主要基团。硫色满酮基位于由LEU-121、TYR-122、MET-508和SER-378等氨基酸残基组成的疏水空穴中, 并形成较强的疏水相互作用, 硫色满酮环中的–C=O基团还能与TYR-118残基形成氢键, 说明–C=O基团在化合物的抗菌活性中也起关键作用。这与阳性对照氟康唑作用模式(图 3)基本相似。活性数据和对接结果表明此类化合物的抑菌作用可能与抑制CYP51有关。
结论本文设计合成了21个未见文献报道的含1, 3, 4-噻二唑片段的硫色满酮衍生物。采用微量稀释法对目标化合物进行抗真菌活性的测试。结果表明化合物5n、5o对R.c.抗菌活性优于阳性对照多菌灵, 最小抑菌浓度均为16 μg·mL-1。化合物5j对C.c.、R.c.、A.n.的抗菌活性也优于阳性对照多菌灵; 化合物5a、5h、5i和5o对A.f.、C.a.的抗菌活性(MIC: 8~32 μg·mL-1)要优于阳性对照氟康唑(MIC: 32~64 μg·mL-1), 其余化合物的抗菌活性较弱。分子对接研究结果表明, 含1, 3, 4-噻二唑片段的硫色满酮衍生物与阳性对照氟康唑的作用模式相似, 说明噻二唑类化合物的抑菌作用可能与抑制CYP51有关。
实验部分1H NMR, 13C NMR和1D-noesy由Bruker AVIII- 600 MHz核磁共振波谱仪完成; HR-MS由Bruker apex ultra 7.0 T傅里叶变换离子回旋共振质谱仪得到; 中间体的MS由Agilent LC/MSD Trap XCT液质联用仪获得; 熔点由SGW X-4显微熔点仪测定。其他试剂和溶剂未经特别说明均为国产或进口分析纯。
1 化合物合成 1.1 化合物2的合成将取代苯硫酚0.1 mol和β-氯丙酸0.12 mol置于500 mL圆底烧瓶中, 加入含0.24 mol氢氧化钠的溶液300 mL, 充分搅拌均匀, 100 ℃反应。TLC跟踪监测, 待反应完全后冷至室温, 用稀盐酸调节pH 1, 析出大量白色沉淀, 抽滤, 大量水洗滤饼, 用乙醇/水重结晶(1:1), 得化合物1a~1c, 收率为72%~85%。化合物的数据与文献[23]一致。
按1 g化合物1用4 mL浓硫酸的比例将化合物1溶于浓硫酸中, 室温放置12 h, 冰解, 析出大量浅黄色固体, 抽滤, 水洗至中性。用50%乙醇重结晶, 得到化合物2a~2c。收率为80%~85%, 化合物的数据与文献[23]一致。
1.2 化合物3的合成取氢化钠0.15 mol溶于甲酸乙酯150 mL中, 用恒压滴液漏斗向其中缓慢滴加0.05 mol化合物2的甲酸乙酯溶液, 30 min滴完, 在冰浴条件下搅拌, 反应12 h。反应液用水萃取3次, 合并水相然后用浓盐酸调节pH 2~3, 静止12 h析出大量沉淀。抽滤水洗得到化合物3a~3c, 产率72%~82%。化合物的数据与文献[24]一致。
1.3 化合物4的合成当R2为甲基、乙基等脂肪族取代基时:取氨基硫脲0.11 mol、有机酸0.11 mol和浓盐酸30 mL加入到50 mL圆底烧瓶中, 搅拌回流3 h, 冷却至室温, 用氢氧化钠调pH 8~9, 冷却析出大量白色沉淀, 抽滤, 水洗得到化合物4a、4b, 产率75%~85%。化合物的数据与文献[25]报道一致。
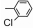
当R2为苯基、对氯苯基等芳香族取代基时:将氨基硫脲0.11 mol和取代苯甲酸0.11 mol先加入到100 mL烧瓶中, 然后在搅拌条件下加入三氯氧磷12 mL, 在80 ℃回流反应2 h, 冷却至室温, 在冰水浴下, 缓缓滴加水15 mL, 0.5 h加毕。然后再在110 ℃回流反应5 h, 冷却, 倒入80 mL冰水中, 搅拌出现沉淀。在冰水浴下用氢氧化钠调pH 8~9, 抽滤, 水洗, 干燥, 得粗品。用DMF和H2O (1:2)混合溶液重结晶得到化合物4c~4g, 产率70%~88%。化合物的数据与文献[26]报道一致。
1.4 目标化合物5a~5u的合成取化合物3 (0.01 mol)、化合物4 (0.01 mol)和浓盐酸0.5 mL加入到20 mL乙醇溶液中, 加热回流7~8 h, TLC监测(乙酸乙酯:石油醚=3:5), 反应结束后冷却至室温, 反应液用200目硅胶色谱(乙酸乙酯:石油醚=1:3)纯化, 即得目标产物5a~5u, 产率在47%~92%。
2 抗真菌活性测试本实验受试动物病原真菌目前保存于河北省药物质量分析控制重点实验室, 植物病原真菌目前保存于河北大学药物化学与分子诊断教育部重点实验室。阳性对照品氟康唑购于山东绿因药业有限公司, 两性霉素B来自大连美仑生物技术有限公司, 阳性对照品多菌灵由江苏泰仓农化有限公司提供。本实验采用微量稀释法分别对4种植物病原真菌和6种动物病原真菌进行活性测试[27]。将目标化合物分别用二甲基亚砜溶解, 配制成样品储备液。采用倍比稀释法稀释样品, 得到一系列质量浓度分别是128、64、32、16、8、4和2μg·mL-1样品溶液。对照品的稀释方法同上。用血球计数法将受试菌的菌悬液浓度配成1×105 CFU·mL-1。将样品溶液100 μL及菌悬液100 μL加到96孔板中(每种菌同时作一空白对照), 混匀后植物病原真菌恒温28 ℃培养3~6天, 动物病原真菌恒温35 ℃培养2~7天, 以无菌生长的最低浓度为最小抑菌浓度(MIC值)。
3 分子对接研究分子对接研究运用Sybyl-X2.0中的Surlex-Dock模块进行分析, 靶标蛋白白色念珠菌的甾醇14α-去甲基化酶(sterol 14α-demethylase, CYP51)的晶体结构(ID: 5TZ1)从蛋白数据库(Protein Data Bank, PDB)中获得。对接时设定对接模式为Surflex-Dock (SFXC), 对受体蛋白进行整体分析、残基修正、加氢加电荷、删除配体等处理。设定距离配体分子5Å范围以内的所有氨基酸残基为活性口袋进行对接叠合。然后基于Sybyl-X2.0软件的Surflex-Dock program模块将化合物和氟康唑分别与CYP51的晶体结构进行对接, 并对配体与受体对接模式进行分析。
| [1] | Berne S, Kovačič L, Sova M, et al. Benzoic acid derivatives with improved antifungal activity: design, synthesis, structure - activity relationship (SAR) and CYP53 docking studies[J]. Bioorg Med Chem, 2015, 23: 4264–4276. DOI:10.1016/j.bmc.2015.06.042 |
| [2] | Imran A, Waseem AW, Amber K, et al. Synthesis and synergistic antifungal activities of a pyrazoline based ligand and its copper (Ⅱ) and nickel (Ⅱ) complexes with conventional antifungals[J]. Microb Pathog, 2012, 53: 66–73. DOI:10.1016/j.micpath.2012.04.005 |
| [3] | Fisher MC, Henk DA, Briggs CJ, et al. Emerging fungal threats to animal, plant and ecosystem health[J]. Nature, 2012, 484: 186–194. DOI:10.1038/nature10947 |
| [4] | Imran A, Hassan Y, Vinay DG, et al. Chiral. Separations of imidazole antifungal drugs on amycoat RP column in HPLC[J]. Chromatographia, 2009, 70: 223–227. DOI:10.1365/s10337-009-1106-z |
| [5] | Crešnar B, Petrič S. Cytochrome P450 enzymes in the fungal kingdom[J]. Biochim Biophys Acta, 2011, 1814: 29–35. DOI:10.1016/j.bbapap.2010.06.020 |
| [6] | Yu X, Nandekar P, Mustafa G, et al. Ligand tunnels in T. brucei and human CYP51: insights for parasite-specific drug design[J]. Biomed Biochim Acta, 2015, 1860: 67–78. |
| [7] | Singh A, Paliwal SK, Sharma M, et al. In silico and in vitro screening to identify structurally diverse non-azole CYP51 inhibitors as potent antifungal agent[J]. J Mol Graphics Modell, 2016, 63: 1–7. DOI:10.1016/j.jmgm.2015.10.014 |
| [8] | Kelly SL, Kelly DE. Microbial cytochromes P450: biodiversity and biotechnology. Where do cytochromes P450 come from, what do they do and what can they do for us?[J]. Phil Trans R Soc B, 2013, 368: 20120476. DOI:10.1098/rstb.2012.0476 |
| [9] | Aboul-Enein HY, Ali I. Enantiomeric resolution of some imidazole antifungal agents on chiralpak WH chiral stationary phase using HPLC[J]. Chromatographia, 2001, 54: 200–202. DOI:10.1007/BF02492245 |
| [10] | Maaroof Z. One-pot synthesis of 1, 3, 4-thiadiazoles using Vilsmeier reagent as a versatile cyclodehydration agent[J]. Tetrahedron, 2017, 73: 1867–1872. DOI:10.1016/j.tet.2017.02.042 |
| [11] | Wesam SA, Rajshekhar K, Mahesh BP, et al. Novel imidazo[2, 1-b]-1, 3, 4-thiadiazoles as promising antifungal agents against clinical isolate of Cryptococcus neoformans[J]. Eur J Med Che, 2015, 95: 514–525. DOI:10.1016/j.ejmech.2015.03.021 |
| [12] | Mahmut G, Halit M, Serdar Ç, et al. Synthesis, characterization, quantum chemical calculations and evaluation of antioxidant properties of 1, 3, 4-thiadiazole derivatives including 2- and 3-methoxy cinnamic acids[J]. J Mol Struct, 2017, 1134: 40–50. DOI:10.1016/j.molstruc.2016.12.041 |
| [13] | Jain AK, Sharma S, Vaidya A, et al. 1, 3, 4-Thiadiazole and its derivatives: a review on recent progress in biological activities[J]. Chem Biol Drug Des, 2013, 81: 557–576. DOI:10.1111/cbdd.2013.81.issue-5 |
| [14] | Lamb DC, Kelly DE, Venkateswarlu K, et al. Generation of a complete, soluble, and catalytically active sterol 14α-demethylase- reductase complex[J]. Biochemistry, 1999, 388: 733–8738. |
| [15] | Banfi E, Scialino G, Zampieri D, et al. Antifungal and antimycobacterial activity of new imidazole and triazole derivatives. A combined experimental and computational approach[J]. J Antimicrob Chemother, 2006, 58: 76–84. DOI:10.1093/jac/dkl182 |
| [16] | Ji HT, Zhang WN, Zhou YJ, et al. A three-dimensional model of lanosterol 14α-demethylase of Candida albicans and its interaction with azole antifungals[J]. J Med Chem, 2000, 43: 2493–2505. DOI:10.1021/jm990589g |
| [17] | Zoumpoulakis P, Camoutsis C, Pairas G, et al. Synthesis of novel sulfonamide-1, 2, 4-triazoles, 1, 3, 4-thiadiazoles and 1, 3, 4- oxadiazoles, as potential antibacterial and antifungal agents. Biological evaluation and conformational analysis studies[J]. Bioorg Med Chem, 2012, 20: 1569–1583. DOI:10.1016/j.bmc.2011.12.031 |
| [18] | Cui XH, Tan J, Zhou M, et al. Design, virtual screening, synthesis and anti-hepatitis B virus of oxime derivaties[J]. Acta Pharm Sin (药学学报), 2016, 51: 1578–1583. |
| [19] | Song YL, Yang T, Dong YF, et al. Facile one-pot synthesis of some novel thiazolylpyrazole derivatives with antifungal activity[J]. Chem Lett, 2014, 43: 134–136. DOI:10.1246/cl.130908 |
| [20] | Song YL, Dong YF, Yang T, et al. Synthesis and pharmacological evaluation of novel bisindolylalkanes analogues[J]. Bioorg Med Chem, 2013, 21: 7624–7627. DOI:10.1016/j.bmc.2013.10.034 |
| [21] | Qi P, Jin YH, Guo C, et al. Synthesis and antifungal activity of 3-bromo-4-sulfur (mannose) derivatives[J]. Chin J Med Chem, 2003, 13: 205–207. |
| [22] | Ramprasad J, Nayak N, Dalimba U. Design of new phenothiazine-thiadiazole hybrids via molecular hybridization approach for the development of potent antitubercular agents[J]. Eur J Med Chem, 2015, 106: 75–84. DOI:10.1016/j.ejmech.2015.10.035 |
| [23] | Xiao LW, Li HZ. Synthesis of β-arylthioethers of propionic acid and thiochromanones under microwave irradiation[J]. Chin J Org Chem (有机化学), 2006, 26: 979–982. |
| [24] | Liu XM, Yang GL, Song YL, et al. Synthesis and antifungal activity of some novel (E)-2, 3-dihydro-3-[(phenylamino) methylene]-4H-1-benzothiopyran-4-ones[J]. Lett Org Chem, 2013, 10: 228–234. DOI:10.2174/1570178611310030016 |
| [25] | Le CG, Ding JH, Yang SJ. Synthesis and application of 5-alkyl-2-amino-1, 3, 4-thiodiazole[J]. Chem World (化学世界), 2002, 7: 366–368. |
| [26] | Niu P, Kang J, Tian X, et al. Synthesis of 2-amino-1, 3, 4- oxadiazoles and 2-amino-1, 3, 4-thiadiazoles via sequential condensation and I2-mediated oxidative C-O/C-S bond formation[J]. J Org Chem, 2015, 80: 1018–1024. DOI:10.1021/jo502518c |
| [27] | Han XY, Li SB, Liang GC, et al. Synthesis and antifungal activities of N-1, 3, 4-thiadiazol-2-yl-4-oxo-thiochroman-2-yl- formamide derivatives[J]. Acta Pharm Sin (药学学报), 2017, 52: 113–119. |