 2018, Vol. 53
2018, Vol. 53

·新药发现与研究实例简析·
新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
米哚妥林是多靶标激酶抑制剂, 尤以对FLT3发生突变的激酶特异性抑制而成就了治疗急性髓性白血病 (AML)。米哚妥林是抗生素星孢菌素的N-苯甲酰衍生物, 经简单的结构变换将选择性不强的天然活性物质改变为选择性强的药物, 其中的重要推手是对AML血细胞中FLT3激酶的分子生物学与功能研究, 解析了激酶突变与AML的关系, 因而聚焦于该突变的激酶, 为已经制备好的活性化合物找到了归宿。 (编者按)
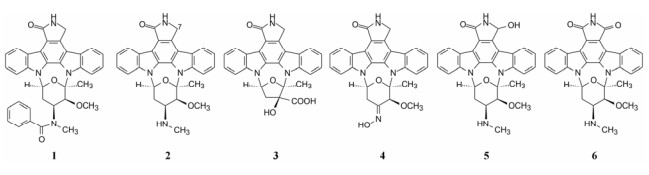
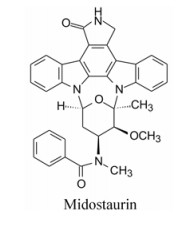
米哚妥林(midostaurin, 1)是2017年美国FDA批准上市的抗肿瘤药物, 由诺华公司(前为汽巴嘉基)研发, 治疗FLT3激酶突变呈阳性的急性髓性白血病和肥大细胞增多症。本品是由天然抗生素星孢菌素(staurosporine, 2)经结构改造而成的。
星孢菌素是Omura等在1977年由链霉菌Streptomyces staurosporeus发酵液得到的抗生素(Omura S, Iwai Y, Hirano A, et al. A new alkaloid AM-2282 of Streptomyces origin. Taxonomy, fermentation, isolation and preliminary characterization. J Antibiot, 1977, 30: 275-281), 其化学结构经单晶X-衍射分析确证为吲哚并吡咯酮并咔唑-N-糖苷的环合物, 为九环并合。最初发现有抗真菌和降血压作用(Rüegg UT, Burgess GM. Staurosporine, K-252 and UCN-01: potent but nonspecific inhibitors of protein kinases. Trends Pharmacol Sci, 1989, 10: 218-220)。直到1986年才发现是蛋白激酶C (PKC)的强效抑制剂(Tamaoki T, Nomoto H, Takahashi I. Staurosporine, potent inhibitor phospholipids/Ca++ dependent protein kinase. Biochem Biophys Res Commun, 1986, 135: 397-402), 由于是多种激酶的泛抑制剂, 且活性强, 因而最初结构改造和成药性的目标是提高对特定PKC激酶的选择性, 但没有聚焦于发生突变的FLT3激酶。在这期间陆续发现了多种含有吲哚并吡咯酮并咔唑为骨架的抗生素, 代表性的有K-252B (3)、TAN-1030A (4)、UCN-01 (5)和RK-1409 (6)。结构的不同在糖基和吡咯酮片段。
2 先导物的优化 2.1 活性评价按照Meyer等所述的方法测定受试化合物对蛋白激酶C (PKC)、依赖于c-AMP蛋白激酶(PKA)、磷酸酶激酶(PPK)、S6激酶(S6-K)、EGFR酪氨酸激酶(PTR)和c-src激酶等的50%抑制浓度IC50 (Meyer T, Regenass U, Fabbro D, et al. A derivative of staurosporine (CGP 41 251) shows selectivity for protein kinase C inhibition and in vitro anti-proliferative as well as in vivo anti-tumor activity. Int J Cancer, 1989, 13: 851-856)。诺华选择星孢菌素(2)为先导物, 是基于它多种激酶有强抑制活性, 从成药性来讲, 这既有利于对肿瘤细胞多靶标的抑制活性, 也有对正常激酶不利的脱靶作用。在没有周边化合物构效关系信息的情况下, 研发者首先对糖环上仲胺作结构变换。
2.2 仲胺的烃基化首先对糖环上的仲胺基用烷基或芳基取代成叔胺, 代表性化合物列于表 1。合成的叔胺化合物大都降低了抑制PKC的活性, 而对其他酶的变化不大或也降低活性, 只有季铵盐化合物9基本保持了对PKC的活性, 但降低了对PKA的活性。叔胺化失去了氮上的氢, 氢键作用或许是与PKC结合的重要因素。季铵化合物的正电荷可能也有利于结合。
|
| Table 1 SAR of alkylated secondary amines on the sugar moiety |

用脂肪酸、芳香酸或氨基酸酰化先导物2糖环上的NH, 代表性化合物列于表 2。仲胺基被酰化后对PKC的抑制活性减弱, 但对其他激酶往往减弱更显著, 因而相对提高了PKC选择性抑制作用。特别是N-苯甲酰星孢菌素(25)与先导物相比, 对PKC活性虽降低了10倍和对PPK降低了15倍, 但对其他激酶活性很弱, 因而选择性优于2。进而合成取代的苯甲酰化合物(26~36), 但选择性没有提高。3, 5-二硝基苯甲酰化合物(33)虽然对PKA和EGFR激酶完全没有活性, 但对PKC的活性降低了20倍。二硝基化合物的成药性也较低。
| Table 2 SAR of acylated secondary amines on the sugar moiety |






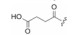


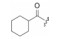
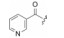




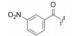

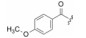

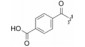
以选择性较强的25为新的起点, 将吡咯烷酮的NH烷基化, 合成的37和38对所有激酶都失去了活性(表 3), 提示该NH是活性的关键基团, 或许是必需的氢键, 或不容有位阻性基团, 因而需保留NH存在。吡咯的内酰胺氧化成酰亚胺40, 仍保持对PKC的活性, 但选择性未曾提升, 而同时做苯甲酰化的44虽然选择性提高了, 但也降低了对PKC的活性。氧化成羟基的两对差向异构体(5, 41~43)对PKC和PKA的活性都有所降低, 而对PPK的活性不降, 这种选择性不可取(Caravatti G, Meyer T, Fredenhagen A, et al. Inhibitory activity and selectivity of staurosporine derivatives towards protein kinase C. Bioorg Med Chem Lett, 1994, 4: 399-404)。
| Table 3 SAR of the modified pyrrolidone compounds |
以上对星孢菌素的结构改造只是为了提高对PKC的活性与选择性, 尚无明确的治疗目标。聚焦于治疗急性髓性白血病(AML)是在发现了AML有30%患者的FLT3激酶发生变异。分子生物学研究表明, 突变后的FLT3的近膜结构域内部串联重复(Nakao M, Yokota S, Iwai T, et al. Internal tandem duplication of the FLT3 gene found in acute myeloid leukemia. Leukemia, 1996, 10: 1911-1918), 还在激酶的活化环套的Asp835残基发生点突变, 导致构象改变, FLT3激酶成为激活的形式, 引起细胞分化不完全并持续增殖(Yamamoto Y, Kiyoi H, Nakano Y, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood, 2001, 97: 2434-2439)。AML的这些分子生物学研究为研发治疗药物提供了靶标支持。
3.2 化合物25抑制变异的FLT3激酶、细胞和体内活性诺华公司系统地评价了上述活性化合物对变异的FLT3激酶和高表达变异酶的细胞—依赖于生长因子的Ba/F3-FLT3-ITD细胞的抑制作用, 发现化合物25对变异的酶和细胞有强效抑制作用, 在24~72 h内抑制Ba/F3-FLT3-ITD细胞的IC50低于10 nmol·L-1, 而对正常的肠Ba/F3细胞在100 nmol·L-1浓度下没有抑制作用, IC50高于500 nmol·L-1, 化合物25的作用是诱导了AML细胞凋亡和终止了细胞周期。
体内动物实验是用骨髓移植表达有FLT3-ITD激酶的逆转录病毒的Balb/c小鼠, 造成模拟FLT3变异的AML动物模型, 从第30天到第88天(实验1)或25~65天(实验2), 每日灌胃给药100 mg·kg-1一次(血药浓度高于IC50), 90天后给药的两组动物全部存活, 而对照组存活率只有20% (均值), 给药组的脾重和白细胞计数都显著低于对照组, 表明化合物25体内抗AML的效果(Weisberg E, Boulton C, Kelly LM, et al. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell, 2002, 1: 433-443)。遂以代号PKC412进入临床研究, 定名为米哚妥林(midostaurin), 证明对FLT3呈阳性的AML和晚期肥大细胞过多症有效, 于2017年批准上市。
|