 2018, Vol. 53
2018, Vol. 53



金黄色葡萄球菌(Staphylococcus aureus)是严重威胁人类健康的主要病原菌之一, 是导致化脓性脓肿的罪魁祸首[1]。耐甲氧西林金黄色葡萄球菌(methicillin-resistant Staphylococcus aureus, MRSA)是院内获得性感染首要致病菌, 可引起社区性大流行[2]; 该类菌株感染可遍及全身并有高发病率及高死亡率特点, 给家庭及社会带来严重的经济负担[3-6]。2017年2月, 世界卫生组织发布了首份急需新型抗生素的重点病原体清单, MRSA被列为第二类重点十分重要栏。我国是MRSA感染大国, 2016年全国细菌耐药检测报告显示:全国平均检出率高达34.4%。MRSA还具有多重耐药性特点, 对临床使用抗生素包括青霉素、头孢菌素、氨基糖苷、四环素、大环内酯、恶唑烷酮以及喹诺酮等均产生不同程度耐药[7-12]。万古霉素(vancomycin)被认为抵御MRSA的“最后一道防线”, 然而, 过去20年间, 中介耐万古霉素金黄色葡萄球菌(vancomycin intermediate-resistant S. aureus, VISA)以及耐万古霉素金黄色葡萄球菌(vancomycin-resistant S. aureus, VRSA)相继被报道[13-15]。2017年7月, G20启动了国际抗生素耐药性十年研发计划。因此, 寻找结构类型新颖、作用机制独特的抗MRSA药物成为全球科学家致力于攻关的科学难题。
本课题组长期致力于从我国生物碱天然产物中寻找化学结构新颖、作用机制独特的创新药物, 由此构建了一个由天然产物衍生、自主创新的化合物库[16-24]。为了寻找结构新颖的抗菌候选物, 对此化合物库开展了表型抑菌高通量筛选, 并幸运地发现了本课题组首先构建的环化小檗碱(CBBR)衍生物——9-乙酰氧基环化小檗碱氯化物1 (图 1), 显示出较强的抗革兰阳性菌活性, 对甲氧西林敏感的金黄色葡萄球菌(methicillin-susceptible S. aureus, MSSA)与MRSA、尤其对VISA均显示较好的抑制活性(表 1), 最小抑菌浓度(minimal inhibitory concentration, MIC)介于1~16 μg·mL-1, 提示其抑菌作用机制可能不同于临床使用抗菌药物。但此化合物中9-乙酰氧基极易被体内酯酶水解, 存在药代不稳定的缺陷。

|
Figure 1 Chemical structure of 1, and its structure modification strategy |
| Table 1 Physical properties and spectra data of all synthesized compounds |
鉴于化合物1独特的化学结构以及抗MRSA活性, 本研究以其为先导物, 以抗菌活性为导向, 针对分子中的E环、B环以及9-位侧链开展了结构修饰与优化, 旨在探讨此类化合物的抗菌构效关系(structure-activity relationship, SAR)的同时, 克服酯基容易被酯酶水解的缺欠, 由此获得抗菌活性强、药代稳定的新型抗MRSA候选物。由此设计合成了14个CBBR衍生物, 包括4个小檗碱衍生物、4个白屈菜红碱衍生物以及6个9-取代CBBR衍生物, 并对代表性化合物开展了体外药代稳定性与初步安全性评价。
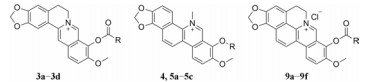
结果与讨论 1 化合物的合成首先, 以市售盐酸小檗碱为起始原料, 在高温负压条件下, 选择性脱除9-位甲基得2, 随后以吡啶为缚酸剂, 与相应的酰氯反应得到9-位酯取代小檗碱衍生物3a~3d (合成路线1)。其次, 以盐酸白屈菜红碱为起始原料, 在高温负压条件下, 选择性脱除甲基得到化合物4, 其与相应的酰氯在三乙胺作用下反应得到7-位酯取代白屈菜红碱化合物5a~5c (合成路线2)。最后, 以小檗碱为原料经还原、取代、环化等反应得到关键中间体CBBR, 其中环化反应为CBBR合成的关键反应。用体积比为1:3的浓盐酸与甲醇代替本课题组之前报道的2%盐酸[23]为环化条件, 不仅时间大为缩短, 而且收率由原来的38%提高到67%。随后高温负压条件下, 对CBBR选择性脱除9-位甲基, 进而在三乙胺作用下, 与相应的酰氯反应得到9-位酯类化合物9a~9f (合成路线3)。其中所合成的白屈菜红碱与CBBR衍生物均为全新结构化合物, 所有目标化合物结构经1H NMR、13C NMR以及HR-MS分析确证。目标化合物的收率、理化参数和波谱数据见表 1。
2 抗阳性菌活性SAR研究

|
Scheme1 Synthesis of compounds 3a-3d. Reagents and conditions: (a) 20-30 mmHg, 195-210 ℃, 40 min; (b) RCOCl, pyridine, CH3CN, 40-91 ℃, 3 h |

|
Scheme2 Synthesis of compounds 5a-5c. Reagents and conditions: (a) 20-30 mmHg, 150-160 ℃, 3 h; (b) RCOCl, triethylamine, CH3CN, 71 ℃, 3 h, 9%-11% (over two steps) |

|
Scheme3 Synthetic route for compounds 9a-9f. Reagents and conditions: (a) NaBH4, 5% NaOH/K2CO3, CH3OH, r.t., 3 h; (b) 40% glyoxal, HOAc/CH3CN, reflux, 6 h; (c) Methanol/HCl (3:1 by vol.), r.t., 24 h, 67% (over two steps); (d) 20-30 mmHg, 195-210 ℃, 60 min; (e) SOCl2, CH2Cl2, reflux, 3 h; (f) Triethylamine, CH3CN, 71 ℃, 3-6 h |
本论文评价了所有目标物对革兰阳性菌, 包括MSSA/MRSA、VISA、甲氧西林敏感/耐药的表皮葡萄球菌(methicillin-susceptible, -resistant Staphylococcus epidermidis, MSSE/MRSE)、耐万古霉素屎肠球菌(vancomycin-resistant Enterococcus faecium, VRE)在内的9株致病菌的体外抑菌活性(表 2)。如图 1所示, 首先, 将先导物1的B环移除获得白屈菜红碱骨架, 由此设计合成了4个白屈菜红碱衍生物4与5a~5c, 它们对所有葡萄球菌活性相当或稍弱于1, MIC介于2~8 μg·mL-1之间; 但对VRE的活性明显提高, MIC在2~16 μg·mL-1之间, 说明移除B环有利于抑菌谱的拓展。
| Table 2 Antibacterial activities of the target compounds against drug-susceptible and drug-resistant gram-positive strains. aThe American Type Culture Collection (ATCC); bStrains isolated from patients in Chinese hospitals |

为了克服先导物酯基极易被酯酶酶解的缺欠, 在9-位引入体积较大的刚性基团以阻碍酯酶的进攻, 分别用3-降金刚烷甲酰基、金刚烷甲酰基、金刚烷乙酰基等取代先导物1结构中体积较小的乙酰基, 获得6个9-位刚性基团CBBR的酯型衍生物9a~9f。其中, 化合物9a、9b对测试菌株抑菌活性普遍优于先导物1, MIC介于0.5~2 μg·mL-1, 特别是对VISA菌株活性是先导物的32倍, 是阳性对照药左氧氟沙星(Lev)的16倍。而9c~9f衍生物的活性均有所降低, 提示:更大取代金刚烷的引入不利于保持活性。
然后, 将先导物1的E环移走由此得到了4个9-酯取代小檗碱衍生物3a~3d, 所得化合物的抑菌活性均完全丧失, MIC > 64 μg·mL-1, 说明E环为活性必需片段。鉴于化合物9a、9b良好的抗MSSA/MRSA活性, 选两者作为代表性化合物进行下一步体外药代稳定性研究。
3 体外药代稳定性评价选择活性较好、含刚性大基团的化合物9a、9b作为代表性化合物进行体外血液药代稳定性测定, 以含有酯键的降血压药物—依那普利马来酸盐(enalapril maleate)作为阳性对照药。取SD大鼠新鲜血液, 并用新鲜的血液孵育所测化合物, 5个不同的时间点取样, LC-MS/MS方法测定目标化合物含量。实验结果如图 2所示, 9a、9b在体外全血孵育半小时后含量分别为48%、9.9%, 均明显优于相应的先导物1的含量0.4%;尤其化合物9a的血液稳定性还优于enalapril (42.6%)。结果提示:化合物9a在体内拥有相对较好的药代稳定性。

|
Figure 2 Structure of enalapril maleate, and the result summary of blood stability assay |
鉴于9a具有良好的体外药代稳定性, 对9a进行了初步安全性评价, 包括对A549细胞的细胞毒及尾静脉急性毒性实验。细胞毒性实验结果如图 3所示:化合物9a对A549细胞株CC50为37.5 μmol·L-1, 结合其MIC值介于0.5~1 μg·mL-1 (1~2 μmol·L-1), 治疗指数介于18.7~37.5之间。一次性给昆明小鼠尾静脉注射(i.v.)化合物9a, 剂量分别为0、20、30和40 mg·kg-1, 密切观察7天。小鼠急性毒性结果显示LD50为30 mg·kg-1, 说明化合物9a还具有相对较好的安全性。

|
Figure 3 Cytotoxic effects of compound 9a on A549 cells. Following pretreatment with compound 9a at the indicated concentrations for 24 h, the cell viability of A549 cells were determined by MTT assay. Control cells were treated with 0.4% (v/v) DMSO |
本课题组前期研究中首次发现全新结构骨架环化小檗碱衍生物1, 具有独特的抗MSSA/MRSA活性, MIC为1~16 μg·mL-1。本研究以1为先导物, 以抗菌活性为导向, 针对分子中的E环、B环以及9-位侧链开展了结构修饰与优化, 共设计合成了14个不同类型的环化小檗碱衍生物, 包括小檗碱与白屈菜红碱衍生物。构效关系分析表明: ① E环为活性必需片段; ②移除B环抗MRSA活性有所降低, 但是抗VRE活性明显优于先导物, 拓宽了抑菌谱; ③ CBBR的9-位引入适当刚性基团有利于提高活性。其中, 化合物9a对标准与临床分离的MSSA/MRSA菌株的活性明显优于先导物1, 尤其对临床难治的MRSA、VISA均显示良好活性, MIC值介于0.5~1 μg·mL-1之间, 提示该类衍生物抑菌的作用机制可能与临床使用抗菌药物不同。同时, 9a还具有较好的体外药代稳定性与安全性特征, 值得进一步研究。研究结果为此类化合物发展成一类新型抗MRSA候选物提供了关键的科学数据。
实验部分熔点用CXM-300型精密熔点仪测定, 温度未校正; 1H NMR和13C NMR用Bruker Avance Ⅲ 500和600核磁共振仪测定, 溶剂为DMSO-d6或CD3OD; HR-MS用Autospec Ultima-TOF质谱测定仪测定; Flash柱分离纯化用Combiflash Rf 200快速制备液相; 荧光检测用ZF-20D暗箱式紫外分析仪; 薄层色谱(TLC)采用E-Merck公司预铺硅胶铝箔卷; 试剂均为分析纯。
1 化学合成 1.1 化合物3a~3d的合成在195~210 ℃负压(20~30 mmHg)下, 将小檗碱(3.71 g, 10 mmol)加热30 min得紫黑色油状物, 经5%盐酸/乙醇酸化, 减压除去溶剂。所得固体以二氯甲烷/甲醇为流动相, 经硅胶柱分离纯化得橙红色固体2 (2.85 g, 80%)。随后将化合物2 (100 mg, 0.28 mmol)溶于无水乙腈(5 mL)中, 加入吡啶(101 μL, 1.26 mmol)和相应酰氯(0.84 mmol), 40~91 ℃反应5~6 h。冷却至固体完全析出, 抽滤, 滤出固体分别用水和二氯甲烷洗涤, 即可得目标化合物3a~3d。
1.2 化合物4与5a~5c的合成在150~160 ℃负压(20~30 mmHg)下, 将白屈菜红碱(3.84 g, 10 mmol)加热3 h得棕色固体, 经5%盐酸/乙醇酸化, 减压除去溶剂。以二氯甲烷/甲醇为流动相, 经硅胶柱分离纯化得目标物4。目标物4 (100 mg, 0.27 mmol)溶于无水乙腈(5 mL)中, 加入三乙胺(170 μL, 1.22 mmol)和相应酰氯(0.81 mmol), 71 ℃反应5~6 h。冷却至固体完全析出, 抽滤, 滤饼分别用水和二氯甲烷洗涤, 得目标化合物5a~5c。
1.3 化合物9a~9f的合成将溶有硼氢化钠(0.83 g, 22 mmol)的5%氢氧化钠溶液10 mL逐滴加入至含小檗碱(7.4 g, 20 mmol)和碳酸钾(8.3 g, 60 mmol)的250 mL甲醇溶液体系中, 室温搅拌3 h, 抽滤收集析出的黄绿色固体, 滤饼依次用蒸馏水(100 mL)和80%乙醇(100 mL)洗涤, 得到黄绿色二氢小檗碱6 (7.1 g, 81%)。将6 (7.1 g, 21 mmol)溶解在160 mL乙腈中, 然后分别加入40 mL乙酸和3 mL 40%乙二醛水溶液, 加热至85~90 ℃回流6 h, 将反应液浓缩, 得到未经纯化的深红色油状物7, 加入体积比为1:3的浓盐酸与甲醇200 mL, 室温搅拌24 h, 将反应液浓缩, 以二氯甲烷/甲醇为流动相, 经硅胶柱分离纯化得橙红色关键中间体CBBR (5.6 g, 67%)。
在195~210 ℃负压(20~30 mmHg)下, 将CBBR (3.95 g, 10 mmol)加热60 min得紫黑色固体。产物经5%盐酸/乙醇酸化, 减压除去溶剂, 以二氯甲烷/甲醇为流动相, 经硅胶柱分离纯化得红色固体8 (3.5 g, 92%)。
1.3.1 方法一:化合物9a、9b和9e~9f的合成氮气保护下, 室温将二氯亚砜(3 mL)滴加到溶有相应羧酸(0.78 mmol)的二氯甲烷(4 mL)溶液中, 48 ℃回流3 h, 冷却、浓缩, 向反应体系中加入5 mL甲苯, 浓缩, 重复操作3次, 得到相应的酰氯不经分离直接用于下步反应。将化合物8 (100 mg, 0.26 mmol)和上步所得的产物混悬于干燥乙腈(5 mL)中, 加入三乙胺(163 μL, 1.17 mmol), 71 ℃反应5~6 h。冷却至固体完全析出, 抽滤, 滤饼分别用水和二氯甲烷洗, 得目标化合物9a、9b、9e~9f。
1.3.2 方法二:化合物9c、9d的合成化合物8 (100 mg, 0.26 mmol)混悬于无水乙腈(5 mL)中, 加入三乙胺(163 μL, 1.17 mmol)和相应酰氯(0.78 mmol), 71 ℃反应5~6 h。冷却至固体完全析出, 抽滤, 滤饼分别用水和二氯甲烷洗, 得目标化合物9c、9d。
2 生物实验 2.1 抗阳性菌活性MIC测定参照CLSI标准, 采用平皿二倍稀释法(agar dilution)进行药敏实验。实验菌用MH肉汤或脑心浸液增菌, 药液用MH肉汤二倍稀释成各种所需浓度, 分别加适量到平皿中, MH琼脂培养基熔化后定量注入含药液的平皿内混匀。样品终浓度分别为64、32……0.06、0.03 μg·mL-1。接种实验菌(接种量为每点1×104 cfu)后置35 ℃恒温培养18 h后观察结果, 无菌生长的平皿中所含药物最小的浓度即为MIC。
2.2 体外药代稳定性实验实验当天取SD大鼠新鲜血液于37 ℃水浴中保温, 选enalapril为阳性对照药。用DMSO将化合物9a、9b和enalapril配置成10 mmol·L-1溶液, 后用45% MeOH/H2O稀释到100 μmol·L-1, 取2 μL稀释后的上述溶液置于98 μL新鲜血液中, 在37 ℃水浴中都分别孵育0、1、5、10、30 min, 到达设置孵育时间后, 迅速加入100 μL水和800 μL孵育终止液(含200 ng·mL-1甲苯磺丁脲和20 ng·mL-1丁螺环酮乙腈溶液), 以4 000 r·min-1离心20 min。取100 μL上清液与200 μL水混合, 振荡10 min, 随后取样用LC-MS/MS分析。
2.3 细胞毒性实验取对数生长期A549细胞制备单细胞悬液, 以每孔6×103个细胞密度接种于96孔板, 每组设置3个复孔, 同时设空白对照(仅加培养液)。待细胞密度达到50%左右, 加入相应的药物作用24 h后, 每孔加入5 mg·mL-1 MTT溶液20 μL, 继续培养4 h。每孔加入DMSO 150 μL, 振荡10 min, 使紫色结晶充分溶解, 酶标仪检测各孔吸光度(A570)值。
2.4 急性毒性实验以昆明种小鼠(18~20 g)为动物模型, 称重后随机分组, 每组10只, 雌雄各半, 分别一次性以0、20、30和40 mg·kg-1的剂量尾静脉注射给药, 密切观察动物7天内的死亡情况, 计算半数致死量(LD50)。
| [1] | Listed N. Classics in infectious diseases. "On abscesses". Alexander Ogston (1844-1929)[J]. Rev Infect Dis, 1984, 6: 122–128. DOI:10.1093/clinids/6.1.122 |
| [2] | Kupferschmidt K. Infectious diseases. Genome study helps contain MRSA outbreak-and breeds new questions[J]. Science, 2012, 338: 1019. DOI:10.1126/science.338.6110.1019 |
| [3] | Roberts S, Chambers S. Diagnosis and management of Staphylococcus aureus infections of the skin and soft tissue[J]. Intern Med J, 2005, 35: S97–S105. DOI:10.1111/imj.2005.35.issue-s2 |
| [4] | Mitchell DH, Howden BP. Diagnosis and management of Staphylococcus aureus bacteraemia[J]. Intern Med J, 2005, 35: S17–S24. DOI:10.1111/imj.2005.35.issue-s2 |
| [5] | Murdoch DR, Corey GR, Hoen B, et al. Clinical presentation, etiology and outcome of infective endocarditis in the 21st century: the international collaboration on endocarditis-prospective cohort study[J]. Arch Intern Med, 2009, 169: 463–473. DOI:10.1001/archinternmed.2008.603 |
| [6] | Lowy FD. Staphylococcus aureus infections[J]. N Engl J Med, 1998, 339: 520–532. DOI:10.1056/NEJM199808203390806 |
| [7] | Levy SB. Multidrug resistance--a sign of the times[J]. N Engl J Med, 1998, 338: 1376–1378. DOI:10.1056/NEJM199805073381909 |
| [8] | Neu HC. The crisis in antibiotic resistance[J]. Science, 1992, 257: 1064–1073. DOI:10.1126/science.257.5073.1064 |
| [9] | Kong KF, Schneper LK. Beta-lactam antibiotics: from antibiosis to resistance and bacteriology[J]. APMIS, 2010, 118: 1–36. |
| [10] | Purrello SM, Garau J, Giamarellos E, et al. Methicillin-resistant Staphylococcus aureus infections: a review of the currently available treatment options[J]. J Glob Antimicrob Resist, 2016, 7: 178–186. DOI:10.1016/j.jgar.2016.07.010 |
| [11] | Auckland C, Teare L, Cooke F, et al. Linezolid-resistant enterococci: report of the first isolates in the United Kingdom[J]. J Antimicrob Chemother, 2002, 50: 743–746. DOI:10.1093/jac/dkf246 |
| [12] | Grundmann H, Airesdesousa M, Boyce J, et al. Emergence and resurgence of meticillin-resistant Staphylococcus aureus as a public-health threat[J]. Lancet, 2006, 368: 874–885. DOI:10.1016/S0140-6736(06)68853-3 |
| [13] | Hidayat LK, Hsu DI, Quist R, et al. High-dose vancomycin therapy for methicillin-resistant Staphylococcus aureus infections: efficacy and toxicity[J]. Arch Intern Med, 2006, 166: 2138–2144. DOI:10.1001/archinte.166.19.2138 |
| [14] | Hiramatsu K, Hanaki H, Ino T, et al. Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility[J]. J Antimicrob Chemother, 1997, 40: 135–136. DOI:10.1093/jac/40.1.135 |
| [15] | Hiramatsu K, Aritaka N, Hanaki H, et al. Dissemination in Japanese hospitals of strains of Staphylococcus aureus heterogeneously resistant to vancomycin[J]. Lancet, 1997, 350: 1670–1673. DOI:10.1016/S0140-6736(97)07324-8 |
| [16] | Li YH, Yang P, Kong WJ, et al. Berberine analogues as a novel class of the low-density-lipoprotein receptor up-regulators: synthesis, structure-activity relationships, and cholesterol-lowering efficacy[J]. J Med Chem, 2009, 52: 492–501. DOI:10.1021/jm801157z |
| [17] | Wang YX, Fu HG, Li YH, et al. Synthesis and biological evaluation of 8-substituted berberine derivatives as novel anti-mycobacterial agents[J]. Acta Pharm Sin B, 2012, 2: 581–587. DOI:10.1016/j.apsb.2012.10.008 |
| [18] | Li YH, Fu HG, Su F, et al. Synthesis and structure-activity relationship of 8-substituted protoberberine derivatives as a novel class of antitubercular agents[J]. Chem Cent J, 2013, 7: 117. DOI:10.1186/1752-153X-7-117 |
| [19] | Li YH, Liu YX, Wang YX, et al. Synthesis and biological evaluation of new 13-n-nonylprotoberberine derivatives as antitubercular agents[J]. Acta Pharm Sin B, 2013, 3: 38–45. DOI:10.1016/j.apsb.2012.12.004 |
| [20] | Yang P, Song DQ, Li YH, et al. Synthesis and structure-activity relationships of berberine analogues as a novel class of low-density-lipoprotein receptor up-regulators[J]. Bioorg Med Chem Lett, 2008, 18: 4675–4677. DOI:10.1016/j.bmcl.2008.07.005 |
| [21] | Yao J, Kong WJ, Jiang JD. Learning from berberine: treating chronic diseases through multiple targets[J]. Sci China Life Sci (中国科学:生命科学), 2015, 58: 854–859. DOI:10.1007/s11427-013-4568-z |
| [22] | Heidarian E, Rafieian-Kopaei M, Khoshdel A, et al. Metabolic effects of berberine on liver phosphatidate phosphohydrolase in rats fed on high lipogenic diet: an additional mechanism for the hypolipidemic effects of berberine[J]. Asian Pac J Trop Biomed, 2014, 4: 429–435. DOI:10.12980/APJTB.4.2014C474 |
| [23] | Bi CW, Zhang CX, Li YB, et al. Synthesis and structure-activity relationship of cycloberberine as anti-cancer agent[J]. Acta Pharm Sin (药学学报), 2013, 48: 1800–1806. |
| [24] | Li YB, Zhao WL, Wang YX, et al. Discovery, synthesis and biological evaluation of cycloprotoberberine derivatives as potential antitumor agents[J]. Eur J Med Chem, 2013, 68: 463–472. DOI:10.1016/j.ejmech.2013.07.026 |