 2018, Vol. 53
2018, Vol. 53

新药发现与研究实例简析
新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
本文是迄今系列刊载的实例简析篇幅最长的一篇, 是因为抗血栓药的特殊要求所致。阿哌沙班的研发者在优化活性、选择性、成药性过程中, 对结构骨架、键连方式和周边基团做了广泛的探索与研究, 在多维层面上进行了优化和构效构代分析。在发表的大量药物化学文章中, 本文是经过取舍后写成的。从苗头化合物到阿哌沙班的上市, 虽然研发的主线明确清晰, 但难以看出苗头−先导物−候选物的路径, “支流多多”, 至为庞杂。研发中结构的构建和变换应用了药物化学许多原理和方法, 诸如骨架迁越、电子等排、片段拼合、分子模拟等。阿哌沙班的研制彰显了新药创制的复杂和难度。
(编者按)
调控血液流动的生理系统精准而复杂, 既要保持在血管内的畅流, 又须在血管受损伤的部位迅速形成血凝而止血。如果血管内形成血栓, 纤维蛋白溶解系统便被活化, 通过消溶血栓, 恢复血流。在正常状态下酶系统呈精准的平衡, 避免栓塞和出血。研究和使用抗血栓药物的宗旨是既可消除或预防血栓形成, 也要避免出血。已有的抗血栓药物如华法林、肝素、水蛭素类似物、“曲班”类药物等, 由于固有的作用环节与机制, 存在不同程度的缺陷, 需要研究更加安全有效的抗血栓药物。
1.2 靶标——Xa因子Xa因子(factor Xa, FXa)是凝血级联反应中的一个蛋白水解酶, 功能是将凝血酶原转变为凝血酶, 它的重要性是内在的机制或外来的刺激下, 引发凝血过程的最后环节。FXa作为酶分子具有放大效应, 一个FXa分子可以将上万个凝血酶原经水解而激活成凝血酶, 因而, 为防止血栓的形成, FXa抑制剂应是更有效率的抗血栓药。
FXa是胰蛋白酶样的蛋白酶, 直接抑制FXa的活性, 抑制凝血酶的生成, 阻断血栓形成和凝血, FXa不影响血小板功能, 因而可降低出血的风险, 在作用机制上优于凝血酶抑制剂和抗血小板聚集药物。
2 苗头化合物 2.1 普筛得到了芳脒化合物杜邦公司在启动研制FXa抑制剂时, 已有多家报道了针对FXa的双芳香脒类抗血栓化合物。杜邦曾经研究过抗血栓形成的糖蛋白IIb/IIIa受体拮抗剂, 是RGD (Arg-Gly-Asp)的模拟物, 考虑到FXa底物凝血酶原的识别位点是EGR (Glu-Gly-Arg), 二者有相似之处, 因而他们筛选了IIb/IIIa受体拮抗剂对FXa的活性, 发现了数个具有弱作用的化合物, 其中化合物1对FXa的Ki值为38.5 μmol·L-1, 虽是含芳脒的化合物, 但只是单边, 化合物拥有知识产权。

|
化合物1是含有异噁唑的芳脒化合物, 以异噁唑苯脒为母核, 将右边的四氢异喹啉丁酮酸片段换成苯脒, “随大流”地设计合成了双芳脒化合物2~5, 考察脒基在苯环的位置与活性的关系。在评价对FXa活性的同时, 测定化合物对凝血酶(越强越好)和胰蛋白酶(越弱越好)的作用, 探究选择性活性。表 1列出了化合物的结构和活性。
| Table 1 Structures of compounds 2−5 |
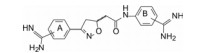
分析构效关系, 提示双苯脒化合物2~5提高了对Xa因子的活性, 其中3的作用最强, 但从选择性上看, 在A环上间位取代的脒基4和5对Xa因子的选择性抑制作用最高。
2.3 异噁唑环上的取代将异噁唑环与酰胺之间的亚甲基除去(降低分子柔性), 以及在异噁唑环上引入含羰基的侧链(R), 合成并评价了化合物6~11, 列于表 2。
| Table 2 Structures of modified isooxazole compounds |
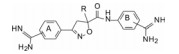
表 2的化合物构效关系分析如下: ① 6减少一个亚甲基, 活性是5的5倍。分子模拟对接到酶的活性部位, 发现A环的间位脒基进入S1腔, 与Asp189形成静电和氢键结合。羰基与Gly318的NH形成较弱的氢键结合(5的羰基不能发生氢键结合)。B环的对位脒基(带正电荷)与Phe174和Tyr99的芳环发生阳离子-π相互作用, 这些信息对于以后的结构设计提供了有意义的指导。②在羰基的α碳上引入含羧基或其甲酯的侧链, 7和8活性进一步提高, 引入的羰基与Tyr99和Gln192形成氢键。而9为甘氨酸甲酯的酰胺, 酯羰基与Tyr99形成强力氢键结合, 因而活性很高。③ 8和10的两个脒基位置调换, 活性和选择性均相近。间位的脒基进入S1腔, 羰基结合的位点则不同。8对FXa有较高的选择性, 静脉灌注大鼠1或5 mg·kg-1·h-1可显著抑制血栓的形成, 而且存在量效关系(Quan ML, Pruitt JR, Ellis CD, et al. Bisbenzamidine isoxazole derivatives as factor Xa inhibitors. Bioorg Med Chem Lett, 1997, 7: 2813-2818)。
3 联苯结构——削减一个苯脒片段 3.1 联苯结构和远端苯环的取代基变换将8作为结构变换的新起点, 用联苯基替换B环的苯脒基, 是为了降低双苯脒的极性, 以利于过膜吸收。12的活性虽然降低了2倍, 但显示只保留一个脒基仍可呈现活性的趋势, 不同于当时研制的双苯脒化合物的潮流。联苯基与FXa的结合腔S4是由Trp215、Tyr99和Phe174等氨基酸残基构成的疏水腔。为此, 合成了取代的联苯化合物, 列于表 3中。
| Table 3 Structures of substituted biphenyl compounds |
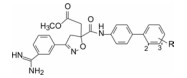
表 3中化合物的构效关系概括如下: ①联苯的2'位连接取代基, 活性高于相应的3'-取代, 是因为分子模拟显示3'-基团与酶活性部位的Lys76和Glu97存在位阻效应的缘故。② 18和22分别为2'-甲氧基和2'-酯基, 活性高于2'-甲基, 推测是氧原子参与形成氢键有利于结合。③ 20和23分别是2'-氨磺酰基和2'-甲磺酰基取代的化合物, 活性最强, 而且20的选择性很高。分子模拟化合物20与FXa的结合特征表明, 磺酰基的氧原子与Tyr99发生强力的氢键结合。图 1是20与FXa活性部位分子对接图。

|
Figure 1 Docking of compound 20 to the FXa active site |
联苯的远端苯环以2'-磺酰基取代为优选, 但尚未探索另一(近端)苯环取代的构效关系, 为此, 固定A环的间位脒基、异噁唑环的乙酸甲酯基, 考察中间的芳环对活性的影响。表 4列出了化合物结构和活性。
| Table 4 Structures of modified ester moiety |
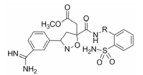
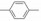
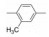
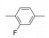
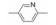


表 4中27和28的活性和选择性(尤其对凝血酶)与20相近或更优, 将这3个化合物拆分后, 比较光活体与消旋体的活性和选择性, 都是(-)对映体活性和选择性优于(+)和(±)化合物。用灌注受试物给家兔动静脉旁路血栓形成模型表明, 这3个(-)优映体抑制血栓形成ID50为0.15~0.26 μmol·kg-1·h-1, 提示有较高的活性(Quan ML, Liauw AY, Ellis CD, et al. Design and synthesis of isoxazoline derivatives as factor Xa inhibitors. J Med Chem, 1999, 42: 2752-2759)。
4 侧链酯基的变换——提高稳定性活性和选择性高的化合物如20的结构中含有酯基, 在体内会发生水解, 在动物的血浆中有相当比例的游离酸存在, 该游离酸对FXa的体内活性显著低于未水解的酯(ID50>4.8 μmol·kg-1·h-1, 20的ID50 = 0.6 μmol·kg-1·h-1)。
为克服该不稳定性质, 对20、27和28的侧链进行了广泛的变换, 包括甲基、烷醚基、烷砜基、烷硫醚基、四唑基等。表 5列出代表性的化合物结构和活性。
| Table 5 Structures of modified ester moiety |
表 5的构效关系提示, 联苯系列去除羧酸酯侧链或用甲基取代(29和30), 活性和选择性变化不大; 3个系列(联苯、嘧啶-苯和吡啶-苯)换成甲氧甲基或乙氧甲基, 也与相应的乙酸甲酯的活性相近, 说明侧链的羰基对于酶结合的贡献不大。换成砜基的活性也未见显著变化, 这个位置对代谢的作用也比较“耐受”, 更换不同的链状片段或基团, 对活性和选择性影响较小。
然而侧链换成四唑甲基, 3个化合物(34、40和45)的活性和选择性都有明显的提高, 进而拆分成光学异构体, 也是(-)-光活体活性高于(±)-消旋体。用家兔研究(-)-34和(-)-40的药代动力学和药效学, 数据列于表 6 (Quan ML, Ellis CD, Liauw AY, et al. Design and synthesis of isoxazoline derivatives as factor Xa inhibitors. J Med Chem, 1999, 42: 2760-2768)。
| Table 6 Pharmacokinetic parameters of 34 and 40 (-)-eutomers |
化合物(-)-40与凝血酶的复合物单晶经X-射线衍射分析三维结构, 表明脒基进入S1腔内, 与Asp189的侧链发生二齿结合; 联芳基处于S2/S3腔中, 末端苯基与Trp215发生“边-面”π-π相互作用; 异噁唑未与酶发生氢键结合; 四唑环靠近Glu192, 因都带有负电荷而相互排斥, 所以只有弱结合作用。晶体结构证明优映体(-)-40为S-构型。(-)-40以同样的分子取向对接到FXa活性部位, 能量优化的四唑环靠近Gln192和Arg143, 他们之间可形成氢键结合。图 2a是(-)-40与凝血酶复合物的晶体衍射图, 图 2b是(-)-40与FXa分子对接图, 相差较小。

|
Figure 2 (a) X-ray crustallography of 40-(-)-eutomer with thrombin; (b) Docking of 40-(-)-eutomer to FXa active site |
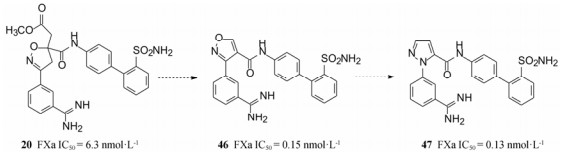
前述化合物的异噁唑啉不是芳香环, 含有手性碳原子, 还证明5-位的碳链对活性影响不大, 因而合成了平面型的异噁唑化合物46, IC50 = 0.15 nmol·L-1, 活性强于20。进而用吡唑替换异噁唑, 47的活性IC50 = 0.13 nmol·L-1, 与46相同。分析20的结构, 异噁唑啉连接出的片段是在3, 5位结合, 间隔一个原子, 而46和47则是邻位相连, 连接位置的区别改变了分子的形状和结合走向, 应是活性提高的结构因素。后经研究47与大鼠胰蛋白酶的晶体衍射表明, 苯脒与联苯片段在吡唑环的邻位连接, 使分子更加刚性, 并有利于远端的苯磺酰胺进入S4腔内, 容易与Trp215发生边对面的π-π相互作用。此外, 47是咪唑的氮原子连接脒基苯, 方便了合成。所以47成为优化结构的新起点。
5.2 甲基吡唑活性更强进而合成了3-甲基取代的吡唑化合物48, 对FXa的抑制活性提高了1个数量级, 结构生物学研究表明, 甲基接近溶剂可及的表面, 与Gly218和Cys220相结合。以47为先导物变换远端苯基和磺酰胺基, 合成的代表性化合物列于表 7中, 以探索化合物结合于S4的片段的构效关系。
| Table 7 Structures of the modified terminal phenyl ring |
表 7中化合物的构效关系表明, 吡唑系列与异噁唑啉的化合物活性水平相当, 在联苯、联吡啶-苯和联嘧啶-苯化合物中, 远端苯环的取代基对活性的贡献依次为SO2CH3>SO2NH2>CF3>H。这个次序也表现在后来的优化中。
|
母核中的3-甲基吡唑是个良好的片段, 但增大烷基取代, 活性则下降, 提示该位置结合的空间不宜是大基团。三氟甲基替换甲基仍保持较高的活性, 表 8列出的代表性化合物, 是含有三氟甲基吡唑的母核, 变换联芳基和磺酰氨基对活性的影响。
| Table 8 Structures of trifluoromethyl pyrrazole moiety |
分析上述化合物的构效关系, 联苯系列的3-三氟甲基与3-甲基的活性相近, 而联嘧啶-苯的3-三氟甲基取代活性强于相应的甲基化合物。62和64整合了所有的优势基团, 因而有最强的活性。
5.4 苯脒-吡唑-联芳基化合物的体内活性对上述高活性和选择性的化合物用静脉注射或灌胃比格犬, 考察药代动力学和药效学性质, 表 9列出的数据表明, 多数化合物有较低的清除率分布容积和较长的半衰期。但口服生物利用度低于5%, 未能达到口服给药的要求。分子结构中含有碱性较强的脒基(pKa~10.7), 在体内分子带有正电荷, 不易过膜吸收, 因而口服吸收差, 这也可从体外Caco细胞的过膜性能差反映出来。
| Table 9 PK and PD parameters of typical compounds |
脒基的强碱性不利于胃肠道吸收, 却是与FXa的Asp189形成盐或氢键的基团。用碱性较弱的苄胺(pKa = 7.8)代替苯脒, 活性略弱于脒基化合物1~2个数量级, 但因脒基具有pM水平的高活性, 经得起活性的消减, 以弥补过膜吸收性, 优化药代+药效的总效果。表 10列出了有代表性化合物的结构与活性。
| Table 10 Structures of benzylamino compounds |
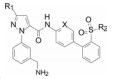
分子模拟方法将72对接于FXa活性部位, 苄氨基与Asp189形成氢键(但不是脒基的二齿型氢键), 而且也未能与Ser180形成氢键。这可能是活性略低的原因。
由于降低了分子的碱性, 增加了过膜吸收性。表 11列出了上述化合物的部分药代动力学参数。
| Table 11 PK parameters of benzylamino compounds |
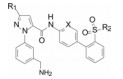
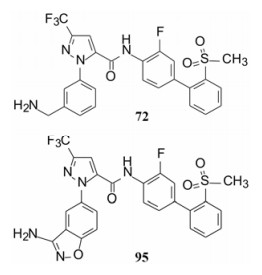
表 11的构-代关系提示, 在苄胺化合物中, 吡唑环上有三氟甲基取代、联苯的近端苯环2位被氟取代、远端苯环4-甲磺酰基取代的化合物72的口服生物利用度最佳, 半衰期长达7.5 h, 用家兔动静脉分流血栓形成模型测定的ID50 = 1.1 μmol·kg-1·h-1, IC50 = 0.15 μmol·L-1。这些较好的活性和药代动力学性质, 使杜邦公司定为候选化合物, 编号为DPC-423, 进入临床前研究(Pinto DJP, Orwat MJ, Wang SG, et al. Discovery of 1-[3-(aminomethyl)phenyl]-N-[3-fluoro-2'-(methylsulfonyl)-[1, 1'-biphenyl]-4-yl]-3-(trifluoromethyl)-1H-pyrazole-5-carboxamide (DPC423), a highly potent, selective, and orally bioavailable inhibitor of blood coagulation factor Xa1. J Med Chem, 2001, 44: 566-578)。
6.2 苯环(或并合环)上其他弱碱性基团的变换在变换成苄胺的同时, 还考虑了碱性更弱的原子或基团的变换, 因为根据离子对相互作用对pKa的扰动, 碱性弱到接近中性的基团也可能与正离子形成氢键或盐(Yamazaki T, Nicholson LK, Torchia DA, et al. NMR and X-ray evidence that the HIV protease catalytic as partyl groups are protonated in the complex formed by the protease and a non-peptide cyclic urea-based inhibitor. J Am Chem Soc, 1994, 116: 10791-10792), 因而合成了苄胺的等排体片段。图 3是联接于N1-吡唑的取代片段, 按照pKa由低到高自左到右顺序排列的。

|
Figure 3 The alkali moieties linked to pyrrazole ring |
设计合成的与吡唑N1相连的脂肪胺和苯胺化合物结构与活性列于表 12。
| Table 12 Structures of aliphatic and aromatic amino groups at pyrrazole ring |

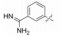


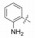
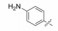
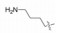

表 12的构效关系提示:苯脒化合物48是活性最强的分子, 但碱性过强而质子化, 吸收差。苄胺65的碱性降低, 活性虽降低200倍, 但仍属高活性。间位苯胺73的碱性很弱, 活性又降低20倍。但邻、对位苯胺(74和75)活性很低, 可能是氨基不能与Asp189结合的缘故。丁胺76和哌啶77也因为链的偏移或环的固定构象都不利于形成氢键结合, 失去活性。
6.2.2 中性的P1片段P1换成碱性更弱乃至中性基团取代的苯环, 代表性的化合物列于表 13中。其中最突出的是对位甲氧基化合物78, Ki = 11 nmol·L-1, 活性只比苯脒低850倍。78是意外筛选发现的并非特意设计合成的化合物, 因为对位氨基活性很低。间位酰胺和间氯化合物79和80有中等活性, 而其余氰基、硫代亚胺和磺酰胺基等化合物(81~84)都没有活性。总之, 对位甲基对活性的贡献是意外独特的。
| Table 13 Structures of neutral moiety at pyrrazole ring |
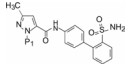
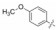

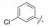
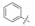
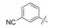

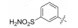
苯脒化合物48与Asp189形成二齿氢键/静电结合是高活性的重要贡献, 为了模拟脒基形成二齿结合的结构特征合成了氨基苯并杂环和有机硼酸等有代表性的化合物列于表 14中。
| Table 14 Structures of bidentate binding moeties |
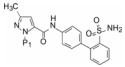

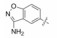
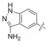
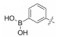

表 14的3-氨基苯并异噁唑(85)有较高的活性Ki = 1.4 nmol·L-1, 其余为中等或无活性, 苯硼酸虽可形成二齿结合, 但因离解成负电荷不利于同Asp189结合。
6.2.4 P1为可形成二齿结合的氨基杂环将碱性基团与形成二齿氢键结合的特征组合在一起, 合成了化合物89~94 (表 15)。
| Table 15 Structures containing basic and bidentate binding groups |
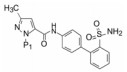

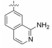



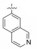
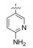
构效关系表明, 其中氨基异喹啉化合物89的活性仍维持在亚纳摩尔的高水平, 对胰蛋白酶的活性很弱, 选择性比苯脒基化合物48高6倍, 可能是由于并环的稳固构象与胰蛋白酶的Ser190 (FXa的相应残基为Ala190)的结合受阻。环内增加氮原子的喹唑啉90和91则活性减弱。93的低活性佐证了碱性和二齿的双重重要性。
结构演化至此(不称其为优化, 是因为结构骨架一直在变换中), 高活性和选择性的化合物89的结构已有相当大的变迁。然而89与FXa复合物的晶体结构分析显示结合的模式没变:进入S1精氨酸识别腔的氨基异喹啉完全质子化(89的pKa值6.7, 在生理pH条件下, 理论上只有13%被质子化), 是由于同Asp189形成离子对相互作用而加大了质子化程度, 还与Gly218形成氢键; 异喹啉环与Ala190有疏水作用; S2/S3处的Gly216与吡唑环上羰基形成氢键; 由Tyr99、Trp215和Phe178的芳环构成的S4疏水腔与远端苯环发生疏水相互作用。图 4a和4b分别是89与FXa活性部位晶体衍射图和结合位点的示意图。

|
Figure 4 (a) X-ray crustallography of 89 with FXa active site; (b) Binding diagrammatic sketch of 89 to FXa active site |
结构演化获得的高活性与高选择性的化合物, 具有里程碑式的意义, 表 16列出了部分高活性化合物的活性、物化、药代参数。
| Table 16 Physicochemical and pharmacokinetic parameters of active compounds |
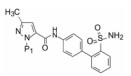
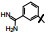

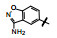


表 16的活性化合物按照pKa值从大到小排列, 从强碱性的脒基到中性的甲氧基, 体外活性逐渐降低, 最大相差800多倍。作为亲脂性量度的脂-水分配系数大致也是由小到大, 亲脂性增加。清除率渐小, 半衰期延长, 由脒基的1.3 h提高到近10 h。口服生物利用度增高, 最大相差12倍(Lam PYS, Clark CG, Li R, et al. Structure-based design of novel guanidine/benzamidine mimics: potent and orally bioavailable factor Xa inhibitors as novel anticoagulants. J Med Chem, 2003, 46: 4405-4418)。从这些趋势性数据可以看出, 优化体外活性要伴随化合物物化性质和体内药代的调整, 选择的候选化合物应兼顾活性和成药性等多重因素, 为了优化某一性质, 往往要对另一性质作出一定的让步或牺牲。
7 联苯远端苯环的变换 7.1 P1为氨基异苯并噁唑化合物作为起点分析构效关系发现进入S1腔内的P1片段, 不仅影响对FXa的抑制活性(与Asp189的结合能力), 而且片段的大小影响选择性作用。化合物72 (代号DPC423)的氨甲基置换为3-氨基苯并异噁唑化合物(95), 活性和选择性显著强于72。然而95的药代性质欠佳。表 17列出了72和95的活性、选择性和药代的参数。
| Table 17 Parameter comparison of compounds 72 and 95 |
化合物95的体外活性的优势与体内药代缺陷之间的矛盾, 解决的方法是保持P1片段不变, 变换远端苯基以优化物化和药代性质。为此, 用杂环置换末端苯环的化合物, 结构和活性列于表 18。
|
| Table 18 Structures of the modified terminal phenyl ring |
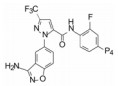

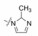
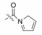
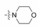
表 18的构效关系分析如下: ① 72的甲磺酰基变换成氨磺酰基, 活性未变, 选择性提高了, 但过膜性显著降低, 系因增加了分子极性的缘故。②远端苯环换成吡啶环, 活性未变, 过膜性仍较低。③用咪唑、四氢吡咯或吗啉置换远端苯环, 增加了助溶基团, 提高了过膜性, 但也因此与血浆蛋白的结合率>97%, 也是活性降低的原因。④ 98的选择性显著低于99, 对凝血酶的抑制作用相差近30倍。因而为调整药代、维持活性强度和选择性, 宜用1-(2-甲基咪唑)基取代远端苯基。
7.2.2 咪唑环上取代基的变换以99为模板, 变换咪唑环上的取代基, 得到102~108, 列于表 19中。异丙基的体积太大, 活性降低, 羟甲基和氨甲基可能因极性强而降低活性, 但106~108的活性、选择性和蛋白结合率优于99, 尤其是107。
| Table 19 Alteration of substituents on imidazole ring |
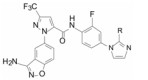

化合物106和107除过膜性的差异较大外, 其余活性参数相近, 进而用比格犬比较了药代动力学性质, 列于表 20。107的口服生物利用度显著高于106, 提示在咪唑环的2位含有二甲胺甲基对活性、选择性和药代性质的优良贡献, 于是将107确定为候选化合物, 进行了全面的临床前评价, 定名为雷扎沙班(razaxaban), 以盐酸盐形式进行临床研究, 口服预防和治疗深度静脉栓塞病, 进入到Ⅱ期临床研究阶段(Quan ML, Lam PYS, Han Q, et al. Discovery of 1-(3'-aminobenzisoxazol-5'-yl)-3-trifluoromethyl-N-[2-fluoro-4-[(2'-dimethylamino-methyl)imidazol-1-yl]phenyl]-1H-pyrazole-5-carboxyamide hydrochloride (razaxaban), a highly potentand selective, orally bioavailable factor Xa inhibitor. J Med Chem, 2005, 48: 1729-1744)。
| Table 20 Pharmacokinetic parameters of compounds 106 and 107 |
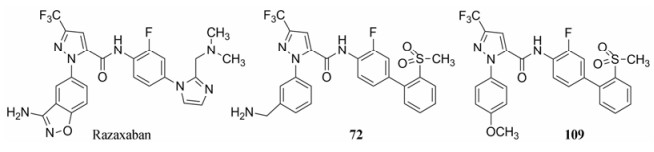
研制的雷扎沙班(107)、72和109等候选物是里程碑式的化合物, 结构中P1片段完全不同, 分别是氨基苯并异噁唑、苄胺和苯甲醚, P4的联芳基也有差异。分子中共有的结构因素是吡唑酰胺。考虑到酰胺键可能在体内被水解成吡唑甲酸和联芳胺, 后者具有潜在的致突变作用的风险, 因而需要避免产生毒性的警戒结构(structural alert), 基于结构变换最小原则, 将酰胺环合到吡唑环上, 形成并合的杂环结构, 以提高代谢稳定性。
8.1 吡唑并嘧啶酮-苯甲醚为母核为了合成方便, 固定远端苯基为苯磺酰胺, 吡唑的3-三氟甲基用3-氰基替换, 制备了含有苯甲醚的嘧啶酮的并合物, 包括中间体110和含氰基化合物111, 氰基的衍生物以及吡唑并吡嗪酮化合物119, 一并列在表 21中。
|
| Table 21 Structures of fused core-biphenylsulfonamides. *aPPT: The compound concentration for activated partial thromboplastin time (aPPT) in vitro clotting assays |
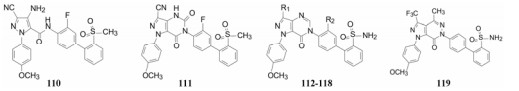
化合物111对FXa的抑制活性与72相近, 但aPPT值很高, 提示吡唑并嘧啶二酮与血浆蛋白的结合率很高。然而吡唑并嘧啶酮化合物112~116对FXa的活性和aPPT活性都较高, 优于111和112, 可与雷扎沙班(107)媲美, 提示苯甲醚连接的吡唑并嘧啶酮母核是有价值的。而且也表明酯基、酰胺基和氰基可以替换三氟甲基仍保持高活性。吡唑并吡嗪酮化合物119也有类似的活性。在吡唑并嘧啶酮母核的基础上, 将雷扎沙班的P4的二甲胺甲基或环状胺基侧链替换磺酰胺基, 合成的化合物120~127列于表 22中。
| Table 22 Structures of tertiary amine at P4 side chain |




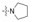
表中化合物的P4部位含有叔胺碱性侧链, 体外仍有较高抑制FXa活性, 羟甲基化合物122活性降低1个数量级, 提示碱性侧链的重要性。吡唑并吡嗪酮化合物128对FXa活性较高, 但aPPT的活性弱, 说明该化合物与血浆蛋白结合率高。
选择高活性化合物评价比格犬的药代动力学性质, 列于表 23中, 可以看出吡唑环以酰胺取代三氟甲基对药代性质有优良的影响, 而氰基没有优势。这为以后的优化提供了新的结构组件。
| Table 23 Pharmacokinetic parameters in dogs of typical compounds |
化合物123和124虽然有良好的药代动力学性质, 但家兔体内实验抗凝血作用低于雷扎沙班3~4倍, 因而不能选定为候选物(Fevig JM, Cacciola J, Buriak Jr J, et al. Preparation of 1-(4-methoxyphenyl)-1H-pyrazolo[4, 3-d]pyrimidin-7(6H)-ones as potent, selective and bioavailable inhibitors of coagulation factor Xa. Bioorg Med Chem, 2006, 16: 3755-3760)。
8.2 吡唑并二氢吡啶酮-氨基苯并异噁唑为母核将吡唑甲酰胺环合成并环以提高代谢稳定性, 表明仍保持较高的活性, 将雷扎沙班的氨基苯并异噁唑与杂环并合吡唑连接, 合成了新的一类化合物, 列于表 24。
| Table 24 Structures of heterocycle-fused pyrrazole compounds |

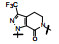
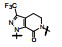
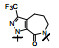
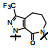

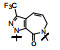

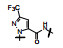
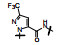
上表的数据表明, 化合物129对FXa的抑制活性强于雷扎沙班, 在近端苯环的邻位连接氟原子(130)活性和体内抗血栓作用都减弱。将吡啶酮扩环成七元环, 无论有无双键都使活性下降。二氢嘧啶酮134对FXa的活性虽比129弱, 但体内抗凝活性强于129。
吡唑并二氢吡啶酮提示是优良的片段, 进而固定该结构对P4部分进行变换, 合成的化合物及其活性列于表 25。
| Table 25 Structures of pyrrazolodihydropyridone moiety |
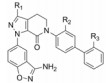
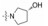
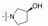
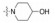
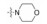



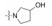
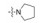
表中化合物的构效关系总结如下: ①变换远端苯环上的叔胺基, 二甲胺甲基(129)和3-R-羟基四氢吡咯(140)活性最强, 体内抗凝作用也最强。②在近端苯环的邻位引入氟原子(146~148), 比相应的化合物活性弱。③ 3-羟基四氢吡咯R构型(140)比S构型(141)强3倍, 体内抗凝活性也强于S构型。④吡唑环上的三氟甲基用甲基代替, 活性降低。
比格犬的药代动力学研究表明, 化合物129和140有良好的性质, 其中140最好。表 26列出了这两个化合物与雷扎沙班的数值(Pinto DJP, Orwat M, Quan ML, et al. 1-[3-Aminobenz-isoxazol-5'-yl]-3-trifluoromethyl-6-[2'-(3-(R)-hydroxy-N-pyrrolidinyl) methyl-[1, 1']-biphen-4-yl]-1, 4, 5, 6-tetrahydropyrazolo-[3, 4-c]-pyridin-7-one (BMS-740808) a highly potent, selective, efficacious and orally bioavailable inhibitor of blood coagulation factor Xa. Bioorg Med Chem Lett, 2006, 16: 4141-4147)。
| Table 26 Pharmacokinetic parameters of typical pyrrazolodihydropiridone compounds |
研究至此, 对P1片段优化, 获得的优势片段有取代的吡唑并二氢吡啶酮、连接苯甲醚、苄胺或氨基苯并异噁唑, P4的优势片段为氨(甲)磺酰联苯和联苯甲基叔胺等。代表性的化合物和候选物为化合物72 (DPC423)、107 (雷扎沙班)、123和140 (BMS740808)等。
下一步是将这些片段作进一步的优化组合, 对体外FXa活性、选择性、体内抗凝作用和药代性质作多维度的优化, 以确定候选化合物。
9.1 苯甲醚-吡唑并二氢吡啶酮-联苯叔胺系列的吡唑环上取代基的变换将苯甲醚、吡唑并二氢吡啶酮、联苯叔胺等优势片段拼接起来, 变换吡唑环上的取代基, 合成的化合物列于表 27中。
| Table 27 Structures of modifying substituent on pyrrazole ring |
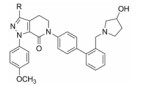
表中的化合物如甲磺酰基、氨甲酰基、氰基和四唑基等与三氟甲基化合物153相比, 都有较高的活性和选择性, 体内抗凝作用显著强于140, 提示这些基团可以移植在不同的母核上。化合物155有良好的药代动力学性质, 较低的清除率(CL = 0.32 L·kg-1·h-1), 半衰期t1/2为5.6 h, 口服生物利用度F = 100%, 因而将氨甲酰基固定, 进一步优化P4片段。
9.2 P4片段优化的化合物固定吡唑环为3-氨酰基, 变换远端苯环的叔胺取代基, 有代表性的化合物的活性、选择性和药代性质列于表 28中。
| Table 28 Stractures of tertiary aminogroup on the terminal phenyl ring |
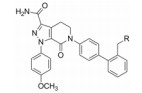
表中所列的代表性的化合物中154仍属优势化合物。
9.3 P4的联苯简化为取代的单苯片段在进行上述结构优化中, 另一个同时研究的是探索P4的变换对活性影响, 发现化合物163的活性和选择性很高(Jia ZJ, Wu Y, Huang W, et al. 1-(2-Naphthyl)-1H-pyrazole-5-carboxamides as potent factor Xa, inhibitors. Part 2: a survey of P4 motifs. Bioorg Med Chem Lett, 2004, 14: 1221-1227), 这个发现很重要, 因为突破了联苯片段, 简化了化学结构。单苯环的对位N-甲基乙酰氨基取代是独特的, 因为游离氨基或其他相关的基团活性都很差, 如表 29所示, 提示酰化的仲胺应是参与结合的重要基团。
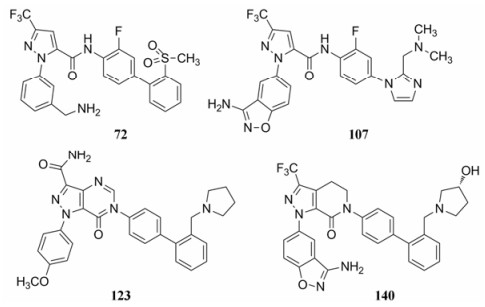
|
| Table 29 Structures of monophenyl compounds |
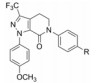


分子模拟考察了化合物163结合于S4腔内的特征, 发现N-甲基与S4腔底部的Trp215发生π-π相互作用, 垂直于苯环的乙酰基与腔内的疏水基团结合。氮上甲基是非常重要的, 因为没有甲基的化合物165活性很弱, 说明只是平面结构的乙酰基不利于结合。环状的内酰胺化合物169和170的活性也较强, 但体内活性弱, 可能是因为脂溶性过强(clog P>7)和较高的血浆蛋白结合率(>99%)的缘故。
9.4 调节脂-水溶性——氨甲酰基取代和候选物的确定为了纠正三氟甲基过强的脂溶性, 将证明有效的基团氨甲酰基连接于163和169吡唑的3位处, 分别得到171和172, 活性列于表 30中。
| Table 30 Comparison of PD, PK and physicochemical parameters between 171 and 172 |
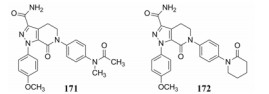
这两个化合物的体外抑制FXa和体内抗凝血作用以及选择性都很强。对人体的蛋白酶和肝脏CYP的活性很弱, 如172与肝微粒体温孵的半衰期>100 min, Caco-2细胞的过膜性也非常高。172的血浆蛋白结合率为87%, 家兔抗血栓的IC50 = 329 nmol·L-1, 口服半衰期为5.8 h, 口服生物利用度F = 58%。遂确定172为候选化合物, 命名为阿哌沙班(apixaban), 经临床前研究和临床试验, 美国FDA于2014年批准百时美施贵宝公司的阿哌沙班上市, 用于预防和治疗深度静脉栓塞和肺栓塞(Pinto DJP, Orwat MJ, Koch S, et al. Discovery of 1-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4, 5, 6, 7-tetrahydro-1H-pyrazolo[3, 4-c]pyridine-3-carboxamide (Apixaban, BMS-562247), a highly potent, selective, efficacious, and orally bioavailable inhibitor of blood coagulation factor Xa. J Med Chem, 2007, 50: 5339-5356)。
9.5 阿哌沙班与Fxa的结合特征阿哌沙班与FXa酶的复合物单晶X-射线衍射表明, 与雷扎沙班的结合模式相似。图 5中深色的分子是阿哌沙班, 黑点构成的虚线代表氢键, 数字为距离Å, 圆点代表水分子。苯甲醚进入S1腔, 甲氧基未显示与特定的基团结合。吡唑环的N2与Gln192的骨架NH形成氢键(3.2Å); 与C3相连的酰胺的羰基与水缔合; NH与Glu146的羰基形成氢键(3.1Å); 母核的内酰胺羰基与Gly216的NH发生氢键结合(2.9Å); 苯基哌啶酮结合于S4腔中, 处于Tyr99与Phe174之间, 与Trp215发生“边与面”的π-π相互作用; 虽然未见哌啶酮的羰基与酶有结合作用(只与水分子结合), 但发生了构象的变化, 取向于Tyr99、Phe174和Trp215构成的空间中。

|
Figure 5 X-ray crystallography of the complex between apixaban and FXa active site |