 2018, Vol. 53
2018, Vol. 53

干扰蛋白-蛋白相互作用的小分子, 批准上市的第一个药物是venetoclax。一些蛋白酶抑制剂或激酶抑制剂虽然也是干预蛋白与蛋白的作用, 但由于反应位点有特定的结构特征、特异的结合腔, 或辅酶的参与, 分子设计有“着力点”, 相对容易实现。Venetoclax干预的两个蛋白相互作用, 是一个广泛而表浅的弱结合作用, 难以确定切入点。本文解析的研发历程, 涉及了高通量筛选, NMR和X-射线衍射揭示微观结构和作用, 以及在确定靶标蛋白的可药性等方面, 有许多值得借鉴之处。
2 作用靶标细胞程序化死亡(凋亡)是机体清除衰老的、受损伤的和无用细胞的首要机制, 与机体正常发育、组织重塑和免疫应答等都起重要作用, 许多疾病的发生是由于凋亡过程的损坏, 例如肿瘤、自身免疫疾病和阿尔茨海默病等。B细胞淋巴瘤(Bcl)蛋白家族中包含有抗凋亡蛋白如Bcl-2和Bcl-xL, 也有促凋亡蛋白如Bak、Bax和Bad, 二者精确地调控表达, 处于平衡状态。一些肿瘤为了避免和逃逸凋亡, 高表达Bcl-2或Bcl-xL, 因而成为研发抗肿瘤药物的靶标, 通过结合Bcl-2或Bcl-xL, 释放促凋亡蛋白如Bak、Bax和Bad的功能, 达到治疗的目的。
3 蛋白结构和结合特征Bcl-2蛋白家族的三维结构有相同的折叠形式:两个疏水性螺旋, 由5~7个两亲性的α螺旋包围, 后者形成抗凋亡蛋白Bcl-xL和Bcl-2的疏水性沟槽, 是结合促凋亡蛋白Bak、Bax和Bad的部位。蛋白-蛋白相互作用的面积广泛(750~1500 2), 表浅且无特征性结合位点, 增加了设计药物的难度。Bak的BH3肽与Bcl-xL的结合面积相对较小, 大约500 2, 而且结合Bcl-xL的位点是较深的疏水沟槽, Bak蛋白的BH3肽为两亲性螺旋, 占据并结合于疏水沟槽, 这些信息为分子设计提供了线索(Petros AM, Nette sheim DG, Wang Y, et al. Rationale for Bcl-XL/Bad peptide complex formation from structure, mutagenesis, and biophysical studies. Protein Sci, 2000, 9: 2528-2534)。
4 评价活性 4.1 Bcl-xL蛋白的制备含有Bcl-xL质粒的细菌在15N标记的氯化铵(唯一氮源)介质中温孵, 制备均一的15N标记Bcl-xL蛋白(去除了过膜螺旋); 15N和13C双标记的Bcl-xL蛋白在15N标记的氯化铵和13C标记的葡萄糖(唯一碳源)介质中制备。用于NMR测定的蛋白浓度为0.5~1.0 mmol·L-1。
4.2 活性测定评价化合物活性是用NMR测定与Bcl-xL蛋白的结合力, 结合性能越强预示活性越高。这种基于NMR的筛选方法是通过对Bcl-xL蛋白与受试物(或无受试物)增敏15N/1H HSQC谱来确定的。通过比对有或无受试物情况下Bcl-xL中特定氨基酸残基的15N二维核磁变化, 确定化合物的结合能力。
荧光偏振检测法测定化合物抑制Bcl-xL活性, 用荧光素标记的BH3肽(来源于Bad蛋白)作为探针(与Bcl-xL结合常数Kd值20 nmol·L-1), 不同浓度的受试物加入到Bcl-xL和探针的混合液中, 用连续的荧光素灯测定偏振光, 计算受试物的Ki为活性值。
高表达Bcl-xL或Bcl-2蛋白的细胞, 用来测定受试物的功能和强度。
5 化合物筛选 5.1 方法采用基于片段的分子设计方法(SAR by NMR), 确定片段及其定位。首先用9 373个化合物筛选Bcl-xL的第一结合位点。化合物的相对分子质量低于210。筛选结果得到49个Kd值低于5 mmol·L-1的苗头化合物。第二结合位点的筛选是在有第一位点的受试物存在下, 筛选相对分子质量低于150的3 472个小分子, 得到24个Kd值低于5 mmol·L-1的苗头化合物。受试物的离解常数Kd值是基于不同浓度与化学位移的变化值求出的。
5.2 第一结合位点的苗头化合物及其构效关系与Bcl-xL第一位点结合的有代表性的化合物1是4'-氟-联苯-4-甲酸骨架的分子, 二维核磁显示苗头化合物引起Bcl-xL疏水沟槽内的Gly94和Gly138的15N化学位移发生变化, 计算得出Kd为300 μmol·L-1。以化合物1为起点, 合成的周边化合物列于表 1。
| Table 1 Binding of biphenyl (naphthalene) compounds |
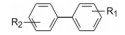
表 1的构效关系提示, 4位羧基是重要基团, 用酚羟基(3)和酯基(4)置换或改为3-羧基(5)都失去活性。4-氟用其他原子或片段替换仍有活性(8~10)。NMR研究还提示, 化合物1结合于Bcl-xL疏水沟槽的中部, 羧基与Arg139发生静电结合, 羧基相当于Bak的Asp83残基。
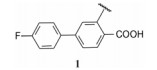
5.3 第二结合位点的苗头化合物及其构效关系联苯甲酸的结合能力弱, 不能阻断Bcl-xL与Bak的结合, 通过比对化合物1与Bcl-xL的复合物与Bak、Bcl-xL复合物的NMR, 发现远处存在第二个结合位点。为发现结合第二位点的苗头分子, 在化合物1的存在下, 用NMR方法筛选了3 500个相对分子质量低于150的小分子。发现结合于第二位点的有代表性分子列于表 2。
| Table 2 Binding activity of fused rings to Bcl-xL |
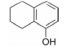
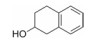


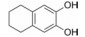
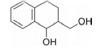
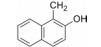
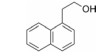
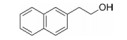
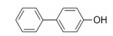
表 2中的化合物活性低于化合物1, 其中化合物11与Bcl-xL的复合物NMR表明, 除1结合第一位点引起Gly94和Gly138变化外, 11结合于第二位点, 导致Gly196的位移变化。两个分离的分子同时结合于Bcl-xL蛋白的不同部位。
5.4 片段的连接 5.4.1 连接位点和连接基的筛选结合于不同位点的两个片段性分子连接成一个分子, 变三元复合物成二元体系, 由于减少了熵损失, 理论上可提高结合能力, 显然, 连接基的长度和取向对结合有重要影响。混合化合物1和11与Bcl-xL三组分的NMR确定了低能量体系下化合物的相对位置, 提示1的羧基邻位是最佳的连接位置, 从而用不同的连接基连接1和第二位点的结合片段, 表 3列出了合成的化合物。
| Table 3 Various fragment at the second binding site and linker connecting to compound 1 |
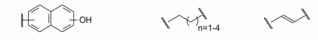
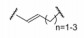
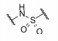
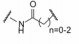

上述化合物经荧光偏振检测, 化合物抑制Bcl-xL活性大多数没有活性(Ki > 10 μmol·L-1), 只有化合物21呈现抑制活性, Ki 1.4 μmol·L-1, 比化合物1强200倍。进而NMR研究21与Bcl-xL的结合特征, 发现乙烯基并非是最佳的连接基。
5.4.2 磺酰胺基的双重作用为了优化连接基的结构与连接方式, 设想用N-酰化的磺酰胺基作为连接基, 氮上有氢的N-酰化的磺酰胺基由于两侧的酰基和磺酰基的拉电子效应有酸性, pKa 3~5, 与羧基相近, 用它连接两个片段, 融合了处于邻位的乙烯基和羧基的作用, 初试的NMR测定证实与Bcl-xL发生结合, 从而合成了包括120个化合物的集中库, 经荧光偏振测试, 发现化合物22抑制Bcl-xL活性Ki值0.245 μmol·L-1, 比21强5倍。图 1是NMR确定的化合物22与Bcl-xL的结合模式, 联苯基处于两个α螺旋之间, Bcl-xL的Phe97区分开两个片段, Phe97的苯基与Tyr194同硝基苯片段发生π-π叠合作用。化合物22可视作3个片段构成:第一片段是联苯基, 第二片段为硝基苯磺酰胺, 第三片段为苯并异硫代吡喃。

|
Figure 1 Binding mode of compound 22 to Bcl-xL by NMR |
确定了N-酰化的磺酰胺为良好的连接基后, 对片段3进行优化, 合成了125个化合物, 其中化合物23为高活性化合物, Ki值36 nmol·L-1。图 2是NMR方法显示的23与Bcl-xL的结合特征, 表明第一结合片段氟代联苯, 第二片段N-酰化的磺酰胺和硝基苯的位置与22相同, 但不同的是第三个片段的硫苯基弯曲回到硝基苯的下方, 此时硫苯基处在蛋白的Phe97与23的硝基苯之间, 而硝基苯在Tyr194下方, 形成层叠的π-π堆积。这些相互作用体现了23的活性强于其他化合物(Petros AM, Dinges J, Augeri DJ, et al. Discovery of a potent inhibitor of the antiapoptotic protein Bcl-xL from NMR and parallel synthesis. J Med Chem, 2006, 49: 656-663)。

|
Figure 2 Binding mode of compound 23 to Bcl-xL by NMR |
化合物23高活性抑制Bcl-xL, Ki值36 nmol·L-1, 但测试介质中若含有1%人血清, 活性下降69倍, 10%血清则完全失活, 提示血清中含有使23失活的成分。后来证明是白蛋白(HSA)的结合, 而α1酸性糖蛋白不影响其结合。进而证明是HAS-Ⅲ的结构域与23的酸性基团N-酰化的磺酰胺相结合, 白蛋白的这个结构域可结合酸性分子和含阴离子的化合物。化合物23与HAS-Ⅲ的Ki < 100 nmol·L-1。
5.5.2 类似物的启示在合成的125个化合物中, 24是23的类似物, 第三片段的苯基多两个甲基。NMR研究表明, 24分别与Bcl-xL和HSA-Ⅲ的结合模式都与23不同。表现在第三和第一片段处: 24的第三片段与HAS-Ⅲ的结合呈伸展形, 苯硫乙基埋入非极性的氨基酸残基中, 提示Bcl-xL和HSA-Ⅲ的末端分别为极性(因此23的苯环向回折曲)和非极性, 因而设想变换乙基为有极性基团可不影响与Bcl-xL的结合, 并促使末端进入溶剂相, 从而削弱与HAS-Ⅲ的结合力, 例如胺、酰胺或砜基不利于与HAS-Ⅲ结合(Hajduk PJ, Mendoza R, Petros AM, et al. Ligand binding to domain-3 of human serum albumin: a chemometric analysis. J Comput. -Aided Mol Des, 2003, 17: 93-102)。23的第一片段分别与Bcl-xL和HAS-Ⅲ相结合的氟代联苯基所处的环境不同, Bcl-xL在氟端尚留有空间, 而且发生部分溶剂化, 而HAS-Ⅲ结合的氟苯基被非极性残基满满地包围, 没有空隙。提示该片段也可加入或变换为极性基团, 以区分与Bcl-xL和HAS-Ⅲ的结合。图 3是化合物23进行分子设计的示意图(Wendt MD, Shen W, Kunzer A, et al. Discovery and structure-activity relationship of antagonists of B-cell lymphoma 2 family proteins with chemopo tentiation activity in vitro and in vivo. J Med Chem, 2006, 49: 1165-1181)。

|
Figure 3 Diagram of 23 bound to Bcl-xL, showing bent-back conformation |
为了消除化合物被HAS-Ⅲ失活的缺陷, 依照上节的分析, 变换氟代联苯基部分, 引入含有极性基团的不同长度的碳链, 合成的化合物列于表 4, 由于之前合成的4'-氟-2'-甲氧基化合物活性略强于23, 故表 3的化合物都含有2'-甲氧基。
| Table 4 Structure and activity of biphenyl tail modified compounds |

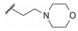
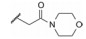
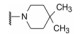
表中的构效关系表明: ①链长为1或2个碳的胺基(酰胺)抑制Bcl-xL活性显著下降; ②链长为三碳的活性与23的活性相当, 推测短侧链未能将胺基“顶出”疏水沟槽进入水相, 而三碳够长; ③表中Bcl-xL+1% HS的数据是在Bcl-xL蛋白中加入1%人血清的受试物活性, 降低的越少, 提示化合物抗失活作用越强。化合物30和32降低人血清的抑制活性; ④叔胺基在介质中易被质子化, 为HAS-Ⅲ所不容, 即使短侧链的25和27也有降低失活作用; ⑤ 31的活性明显高于29, 抑制HAS-Ⅲ的去活作用也强, 是系列中优质化合物。从31和33的数据可以判断, 第一位点引入极性基团可以将活性与失活性(HAS-Ⅲ)分开, 也证明了前述的NMR研究得出的该区域有空闲空间的推断。
与此同时还合成了R为取代的哌嗪化合物, 虽然降低了HAS-Ⅲ的失活能力, 但抑制Bcl-xL的活性也显著降低。然而4', 4'-二甲基哌啶化合物34的活性和抑制HAS-Ⅲ的结合作用都强于23, 因而也成为一个优化切入点。
5.5.4 另一端的变换以化合物31的丙基吗啉或34的4', 4'-二甲基哌啶为固定基团, 变换片段3的结构, 即在乙硫基的α位连接含胺基的侧链, 以提高抑制Bcl-xL的活性和降低HAS-Ⅲ的去活作用。由于α碳为手性原子, 可形成对映体。表 5列出了化合物的结构与活性。
| Table 5 Structure and activity of combination substitution. *Mouse FL5.12 cells transfected with Bcl-xL |
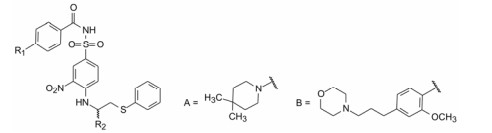
表 5的构效关系提示: ① α碳连接碱性侧链的构型对活性影响显著, R构型的活性强于S构型, 提示R构型引出的侧链有利于结合于Bcl-xL的第三位点, 活性可高达1 nmol·L-1。而S构型的方向使侧链进入水相, 不发生结合。② A和B两种片段如31和34一样, 都显示对抗HAS-Ⅲ的失活作用。③酰胺化合物40和46以及含吗啉的39R和45抑制HAS-Ⅲ的失活作用很弱, 是由于不能被质子化, 有利于结合HAS-Ⅲ。④ 35R和41R在细胞模型上呈现高活性, 即使加入胎牛血清活性仍优于其他化合物, 与HAS-Ⅲ结合很弱, Ki值分别为13.6和94 μmol·L-1, 佐证了对HAS-Ⅲ的抗性。
5.6 里程碑式化合物具有高抑制Bcl-xL活性和高对抗HAS-Ⅲ作用的化合物35R进一步证实可促进放射治疗或紫杉醇引起非小细胞肺癌(高表达抗凋亡蛋白Bcl-xL)的死亡, 表明有促凋亡作用。小鼠移植对多种细胞毒药物无效的人肿瘤A549细胞, 用35R+紫杉醇给药, 抑制率达60%~70%, 且未见增加毒性。由此体内外功能性实验表明35R是BH3蛋白的小分子模拟物。

|
Figure |
化合物35R对多种人癌细胞的抑制效果并不高。由于35R是基于Bcl-xL结构设计的, 没有考虑对Bcl-2蛋白的抑制, 对人体多种高表达Bcl-2的肿瘤高抑制作用很弱, 不能阻止Bcl-2蛋白的抗凋亡作用, 所以抑瘤谱窄。项目研究至此, 意识到当初对靶标的可药性(druggability)的认识有局限性。
Bcl-xL与Bcl-2蛋白序列的同源性虽然只有49%, 其三维结构却很相似(Petros AM, Olejniczak ET, Fesik SW. Structural biology of the Bcl-2 family of proteins. Biochim Biophys Acta, Mol Cell Res, 2004, 1644: 83-94), 例如两个蛋白都有疏水型沟槽, 是结合促凋亡蛋白的BH3结构域的位置, 沟槽的取向与定位都处于两个α疏水螺旋的中间, 只是Bcl-2的沟槽较宽和深, 这个区别为继续修饰结构提供了着力点。为了提高抗肿瘤活性, 设定的新目标是对Bcl-xL/ Bcl-2双靶标作用, 评价化合物的活性分别用两种蛋白作荧光偏振检测。
6.2 契机——化合物47的启示47是变换片段1时所合成的化合物, NMR研究47与Bcl-xL的结合, 发现片段1的苯乙基呈伸展型构象结合于疏水沟槽; 研究与Bcl-2的结合模式, 发现该苯乙基深入到疏水沟槽的深部, 埋入疏水腔中。这为设计双靶标抑制剂提供了修饰位置。图 4是47经NMR研究确定的与Bcl-xL (a)和Bcl-2 (b)的结合模式。然而47对Bcl-xL/Bcl-2的活性不强, 还不够作为先导物进行优化。

|
Figure 4 (a) NMR-derived structure of 47 bound to Bcl-xL (PDB code 2O2M); (b) NMR-derived structure of 47 bound to Bcl-2 (PDB code 2O2F) |
化合物35R的4', 4'-二甲基哌啶是引长疏水链的位置, 因为二甲基作为“把手”可进行基团变化或引长。为广泛探索首先设计了8种片段, 以发现双活性化合物的设计方向。表 6列出了取代的哌啶和哌嗪片段的有代表性化合物活性。
| Table 6 SAR of various 4-substituted piperidine and piperazine structural scaffolds |
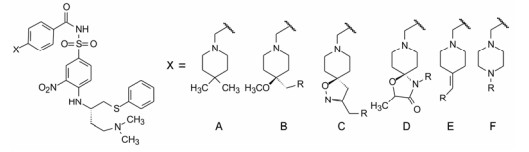
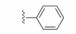
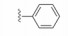
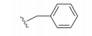
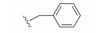
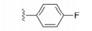
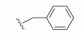
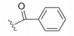
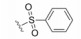
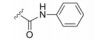
化合物35R哌啶环的4位两个甲基换作不同基团, 一个疏水性基团与疏水沟槽结合, 另一个极性基团进入溶剂相, 4位连接甲氧基和苄基的48, 对Bcl-xL(蛋白和高表达细胞)的活性与35R相同, 但提高了抑制Bcl-2 (蛋白和细胞)活性。异噁唑49和50都提高了抑制Bcl-2活性。亚苄基化合物52对两种蛋白和细胞的抑制作用都很强。
哌啶环换成哌嗪的化合物虽然方便于合成, 但除化合物53 (N-苄基哌嗪)对两种蛋白和细胞有中等活性外, 酰基取代的54~56活性都差。基于这些结果, 下一步的研究集中于B、E和F片段的变换。
6.3.2 确定哌嗪构成的骨架化合物48的R基作烷基、取代苯基和联苯基等变换, 测定对Bcl-xL和Bcl-xL以及高表达的细胞抑制活性, 发现57为高活性化合物。对52的R基用取代苯基和吡啶基连接, 发现化合物58为高活性化合物。对N-苄基哌嗪的苯环作不同的取代, 发现59为高活性化合物。这3个化合物对双靶标的活性列于表 7。
| Table 7 Structure and activity of typical compounds |
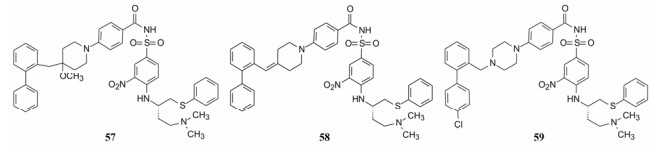
3个化合物的片段1具有共同特点, 都连接了联苯结构, 延伸了疏水性, 深入到Bcl-2蛋白的疏水腔穴中(结合图省略), 也不影响与Bcl-xL的结合。化合物59活性显著强于57和58, 用3株高表达Bcl-2蛋白的滤泡性淋巴瘤细胞评价59活性, 即使含有3%胎牛血清, IC50也低于1 μmol·L-1。移植滤泡性淋巴瘤细胞的小鼠用59、依托泊苷和59加依托泊苷实验, 表明单独应用59的抑制作用相当于依托泊苷的最大耐受剂量, 联合用药可达到90%的抑制率(Bruncko M, Oost TK, Belli BA, et al. Studies leading to potent, dual inhibitors of Bcl-2 and Bcl-xL. J Med Chem, 2007, 50: 641-662)。59进入了临床研究(ABT-737)。但由于水溶性很低, 静脉用药有很大困难, 口服的吸收性因人波动性很大, 显示出59的物化和药代性质有缺陷。
7 优化药代动力学性质进一步优化目标是改善药代性质。为了不影响与靶标蛋白的结合强度, 主体结构不作重大变化, 在非药效团部位加以改动, 评价活性也因此以细胞模型为主, 包括Bcl-2和Bcl-xL依赖性的人小细胞肺癌(H146)细胞模型。
59的相对分子质量799.40, 含有5个苯环、1个脂环、11个氢键接受体、2个氢键给体。分子尺寸大, 脂溶性强, 虽然不大符合Lipinski的口服类药5规则(ROF), 而作为蛋白-蛋白相互作用的抑制剂却是需要的。ROF是小分子药物大数据的统计概率, 不能成为药物设计的羁绊。59的代谢位点是N-去甲基化, 二甲胺基也是结构改造的位点。
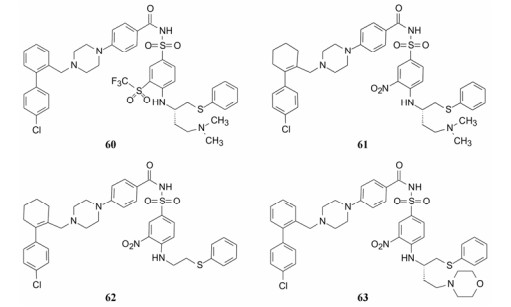
7.1 变换硝基硝基拉电子性有利于提高磺酰胺的酸性, 有助于结合, 但不利于溶解性。换成其他拉电子基团如氰基、三氟甲基、三氟乙酰基和甲磺酰基等化合物的活性都有下降, 其中三氟甲磺酰基化合物60活性较好, 也增加了生物利用度和体内暴露量。
7.2 变换联苯基联苯基刚性过强, 不利于吸收, 将中间的苯环用不同大小的环烯烃替换, 以保持片段1的构象的同时, 改善物化性质。结果表明都有一定的细胞活性, 环的大小对活性影响很大, 其中环己烯化合物61活性最强。
7.3 变换二甲胺乙基侧链去除二甲胺乙基侧链的化合物62的口服生物利用度为28%, 提高了37倍, 但细胞活性显著下降, 增加了与血清的结合, 因为设计这个侧链本意就是抑制与血清白蛋白的结合。用吗啉环(pKa ≈ 7.5)代替二甲胺基, 化合物63的细胞活性仍在亚微摩尔水平, 生物利用度提高了4倍(F = 16%), 所以吗啉环是维持活性提高吸收性的基团, 可与其他优化基团搭配。表 8列出了这些化合物的细胞活性、药代性质与优化前的59的比较。
| Table 8 Activity and PK properties of typical compounds |
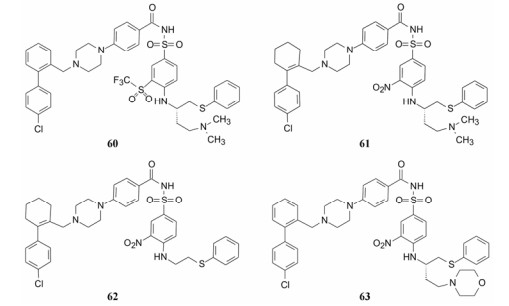
分析表中化合物的不同位置优化的构效(SAR)和构代关系(SPR), 曲线下面积(AUC, 代表一定时程内的药物暴露量)与药效的比值(AUC/EC50)可表征化合物的药代和药效的总体质量, 60和61显著提高了活性, AUC也优于化合物59, 提示三氟甲磺酰基和环己烯分别替换硝基和苯环是优势选择; 62没有碱性侧链, 活性很差, 说明62是不可取的; 63的吗啉基对药代呈正贡献。
7.4 优势片段的组合将片段1换成4-氯代苯基环己烯, 片段2的硝基换成三氟甲磺酰基, 片段3换作吗琳基, 拼合成新的分子, 化合物64和65是有代表性的化合物, 药效学和药代动力学性质如表 9所示。
| Table 9 PD and PK of compound 64 and 65 |
表中数据表明, 化合物64和65由于整合了环己烯(替换苯环)、三氟甲磺酰基(替换硝基)、吗啉(替换二甲胺基)诸因素, 几乎是加和性地改善了药代性质, 比59提高了20~30倍, 药理活性也有所提高。由于蛋白-蛋白相互作用的热域(hot sots)具有播散性, 热域之间独立存在, 相互影响较小, 因而这种加和性(药效和药代)比较直观明显, 与作用于酶或受体的药物(基团间相互影响显著)有所不同。用小鼠、大鼠、犬和猴系统地比较了64和65, 绝对生物利用度都达到20%, 65优于64。进而用小鼠多种移植性肿瘤模型灌胃65, 表明有抑制作用。65的代号为ABT-263, 定名navitoclax, 确定为候选化合物, 进入临床试验研究(Park CM, Bruncko M, Adickes J, et al. Discovery of an orally bioavailable small molecule inhibitor of prosurvival B-cell lymphoma 2 proteins. J Med Chem, 2008, 51: 6902-6915)。
8 结构再改造—消除抑制血小板的不良反应 8.1 降低血小板的不良反应Navitoclax (65)的Ⅱ期临床试验, 显示对患者有抗肿瘤作用, 但同时出现血液毒性, 与临床前实验发现剂量依赖性的降低血小板相吻合。研究表明是由于抑制Bcl-xL蛋白的缘故。这个不良反应限制了给药剂量, 致使治疗窗口狭窄。这个结果是对靶标蛋白Bcl-xL的进一步质疑, 也由此可见首创性药物靶标风险时刻存在, 靶标的可药性被不断地考量。因而拟从化学结构上改造, 去除对Bcl-xL抑制作用, 保留和提高抑制Bcl-2的活性。
8.2 分析结合特征促凋亡的Bcl-xL和Bcl-2蛋白与抗凋亡蛋白的BH3结构域的结合模式非常相似, 这是分开两种活性的困难所在, 但有必要深入分析结合的微观特征, 为提高选择性修饰分子结构提供信息。
Bcl-xL与Bcl-2共同的结构域是两个疏水性α螺旋被5~7个两亲性的α螺旋围绕, 其中4个螺旋形成了长度为20疏水沟槽, 与抗凋亡蛋白的BH3多肽域结合。丙氨酸扫描提示, Bcl-xL和Bcl-2沟槽中主要结合位点是P2和P4疏水腔, 以及精氨酸与BH3的天冬氨酸残基的静电结合。
Navitoclax与Bcl-2复合物晶体结构显示, 苯硫基进入P4疏水腔中, 还与磺酰胺发生π-π叠合作用。4-氯代苯基环己烯片段结合于P2疏水腔。
8.3 结构变换 8.3.1 去除苯硫基侧链上述分析并不能分辨Bcl-xL与Bcl-2的结构差异, 因而通过变换小分子结构, 分析构效关系方法, 即药物化学的试错法(trial and error), 提高对Bcl-2的选择性作用。通过系统地除去或变换重要的结合基团, 发现去除苯硫基的化合物66对Bcl-2失去了部分活性(Ki = 59 nmol·L-1), 但明显降低了抑制Bcl-xL作用(Ki = 5 540 nmol·L-1), 提示有可能区分两个靶标蛋白。
化合物66与Bcl-2的晶体结构显示结合模式与navitoclax相似, 但片段3占据的P4腔穴的空间变小。另一个特征是66与Bcl-2二聚体结合, 第2个Bcl-2蛋白的色氨酸残基(Trp30)嵌入到66结合的P4腔内, 吲哚环与硝基苯形成π-π叠合作用, 与navitoclax的苯硫基的π-π叠合相似。Trp30的吲哚氮原子与Bcl-2的Asp103发生氢键结合(Bcl-xL的残基为Glu103)。图 5是66与Bcl-2二聚体的晶体图, 紫色的吲哚环与硝基苯发生π-π叠合, 氮原子与Asp103发生氢键结合。

|
Figure 5 Crystal diagram of the complex of 66 with Bcl-2 |
模拟上述的结合特征将吲哚环经醚键连接在母核苯环上, 化合物67结合Bcl-2有高度选择性, Ki < 0.1 nmol·L-1, 与Bcl-xL结合的Ki > 660 nmol·L-1, 活性相差千倍。与Bcl-2二聚体的晶体图(图 6)显示吲哚环处于Trp30的位置, 氮原子与Asp103发生氢键结合, 此外, 吲哚的并合苯环与Asp107的距离适于氢键结合, 提示可利用该位置换作氮杂吲哚以增强结合作用。

|
Figure 6 Crystal diagram of the complex of 67 with Bcl-2 |
优化至此, 将原来作用于双靶标蛋白的navitoclax改造成只选择性结合于Bcl-2的化合物。整合的结构因素包括有利于药代性质、不与血浆白蛋白结合、增强对Bcl-2结合和消除对Bcl-xL的作用等结构因素, 经药物化学和构效关系的反馈, 优化出化合物68 (ABT-199)。

|
Figure |
68选择性抑制Bcl-2, 而对Bcl-xL作用很弱, 表 10列出了对靶标蛋白和高表达细胞的作用。例如对Bcl-2高表达的急性淋巴白血病细胞(ALL) EC50 = 8 nmol·L-1, 而对Bcl-xL高表达的H146细胞EC50 > 4 000 nmol·L-1。68消除了抑制血小板的不良反应, 小鼠灌胃100 mg·kg-1 (AUC = 2261 µg·h·mL-1), 血小板计数未见变化, 而navitoclax犬口服5 mg·kg-1 (AUC = 115 µg·h·mL-1), 用药后6 h的血小板降低95%, 是由于作用靶标不同的缘故。
| Table 10 The selective action of 68 to BCL-2 |
化合物68可口服吸收, 6~8 h血药浓度达峰, 半衰期26小时。定为候选化合物, 名为venetoclax, 经临床实验, 证明对17号短臂染色体缺失的慢性淋巴白血病有效, 于2016年4月FDA批准上市(Souers AJ, Leverson1 JD, Boghaert ER, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitu mor activity while sparing platelets. Nat Med, 2013, 19: 202-208)。
9 后记Venetoclax是全球第一个针对蛋白-蛋白相互作用的小分子抑制剂, 屡改靶标和先导物, 研发历程20年, 彰显出首创性药物的风险与艰辛。由开始以Bcl-xL为靶标, 中间改换为作用于Bcl-xL和Bcl-2双靶标, 后来才聚焦于Bcl-2靶标, 是在确定了候选物乃至进入临床试验后的更换, 提示确证药物靶标贯穿于研发的全过程, 风险贯穿始终。
Venetoclax是基于片段的药物发现(FBDD)的成功范例, 用SAR by NMR方法和片段连接策略构建苗头化合物, 在由苗头过渡到先导物、优化活性、提高选择性、消除脱靶作用等过程中, NMR和X-射线衍射分析微观结构起到指导作用, 分子模拟和构效关系则是验证与反馈的重要手段。Venetoclax的成功也体现了概念验证是至关重要的。此外, venetoclax的结构和物化性质几乎完全突破了类药5规则, 创新不能墨守成规, 因为指望相对分子质量在500以下的小分子阻断两个蛋白的结合是难以实现的。