 2018, Vol. 53
2018, Vol. 53



在药物发现过程中, 先导化合物的代谢稳定性(包括肝代谢稳定性和血浆稳定性等)是影响其成药性的关键因素[1-3]。在药物研发案例中, 很多先导化合物都存在代谢稳定性差的问题, 而很多具有良好药理活性的化合物由于不够稳定最终导致研发失败终止。因此, 改善化合物的代谢稳定性是在寻找候选药物过程中亟待解决的重要问题。尽管化合物的肝代谢稳定性被普遍认为是药物发现过程中所面临的最主要的挑战之一[4], 化合物的血浆稳定性仍然是新药研发过程中一个重要的影响因素[5-7]。一些含有特定官能团的化合物在血液中易分解, 而不稳定的化合物常常具有较高的清除率和较短的半衰期, 从而导致体内药动学和药效学性质不佳。如果在新药研发过程中只关注化合物的肝代谢稳定性而忽视其血浆稳定性, 就可能无法有效地解决化合物的体内稳定性问题。因为肝脏中的代谢酶和血液中的酶不同, 在体外稳定性评估中, 化合物在肝微粒体中稳定并不能代表它在血浆中也同样稳定。因此, 提高先导化合物的血浆稳定性对于新药研发同样具有重要意义。通过对先导化合物进行结构优化[8-13], 提高化合物的血浆稳定性、延长药物在体内的作用时间、增加体内的暴露量、降低化合物的清除率、提高生物利用度, 进而改善其药动学和药效学性质对于先导化合物结构优化十分必要。本文主要介绍了通过化学结构修饰提高化合物血浆稳定性的策略, 包括:生物电子等排、增加空间位阻、成环修饰以及骨架跃迁等。通过总结提高化合物的血浆稳定性的结构修饰策略, 以期能够为候选药物的设计与开发提供参考。
1 血浆稳定性的基本原理血浆是血液的重要组成部分, 其主要成分是水、血浆蛋白、葡萄糖、激素、矿物离子以及多种水解酶等。血浆中的水解酶主要有胆碱酯酶、脱氢肽酶、脂肪酶和磷酸酶等。血浆和肝脏中的水解酶是存在差异的。例如假性胆碱酯酶(pseudocholinesterase)由肝脏合成, 但主要存在于血浆中, 可水解乙酰胆碱和其他胆碱类化合物[10]。血浆中还存在淀粉酶和脂肪酶, 其主要来源于胰腺和唾液腺等, 淀粉酶和脂肪酶对多糖类及脂肪类化合物的水解起重要作用, 而肝脏中则不含该类水解酶。醛缩酶B (aldolase B)在糖酵解和糖异生过程中具有重要作用, 其主要存在于骨骼肌和肝脏中, 而在血浆中的含量较低[11]。磷酸二酯酶3B (phosphodiesterase 3B)的主要作用是参与磷酸脂类的水解, 其主要存在于脂肪组织和肝脏中, 在血浆中的含量较少[12]。血浆中的各种水解酶的含量和活性取决于个体的种族、性别、年龄和健康状况等[14]。当小分子化合物与血浆中的某种水解酶具有一定亲和力的时候, 其结构中特定的官能团就会被水解酶水解, 从而导致具有生物活性的化合物在血浆中的浓度迅速降低, 以致无法发挥有效的治疗作用。在选择活性化合物开展体内药效学评价时, 血浆稳定性数据可以直观地反映出化合物可能会出现的稳定性问题, 这将有助于研究人员挑选稳定性更好的化合物进行后续研究[6]。
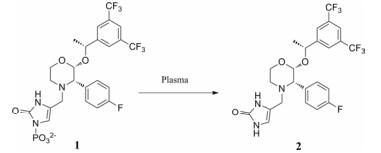
在血浆中容易被降解的官能团主要有酯基、酰胺、内酯、氨基甲酸酯、内酰胺、磺酰胺以及肽类结构等[6]。由于对映异构体与血浆中水解酶的亲和力差异, 化合物的血浆稳定性也受到手性因素的影响[15]。另外, 化合物在不同种属之间的血浆稳定性有时也存在较大差异, 这可能与不同种属中水解酶的活性、含量以及结构差异有关。如表 1所示, 化合物1是人神经激肽-1 (hNK-1)受体拮抗剂的磷酸酯前药, 其在血浆中的稳定性顺序如下:大鼠<犬<人[16]。一般情况下, 化合物在啮齿动物体内的稳定性要低于人体内的稳定性。因此, 在药物研发的初期阶段, 有必要对先导化合物的血浆稳定性进行评估, 尽早发现潜在的不稳定结构并对其进行必要的结构修饰, 或者挑选稳定性更佳的先导化合物开展进一步的药理活性和代谢性质的优化[6]。
| Table 1 The plasma stability of compounds in different species |
通常情况下, 提高化合物在血浆中的稳定性, 从而增加化合物在体内的暴露量以及延长药效作用时间等是研发人员进行先导化合物类药性优化的主要目标。但是, 血浆的降解作用也并非有弊无益, 药物化学家可以利用它的降解特性来进行前药和软药的设计优化。为了提高化合物的渗透性和代谢稳定性, 可以采用前药策略[17-19]增加进入血液的前药浓度, 使得前药在血液中水解并释放出活性成分。另一种利用血浆降解特性的策略是设计软药。软药是指容易代谢失活的药物, 使药物在完成治疗作用后, 按预先设定的代谢途径和可以控制的速率分解、失活并迅速排出体外, 从而避免药物的蓄积毒性, 这类药物被称为软药。软药和前药相反[17, 20], 软药在局部使用时是有药理活性的, 但进入血液后会迅速降解成非活性成分。早期开发的糖皮质激素在长期使用后会出现较多的不良反应, 例如:诱发或加重感染、引起物质代谢和水盐代谢紊乱、诱发高血压和动脉粥样硬化、白内障和青光眼等。因此, 人们迫切需要研发一类安全有效的糖皮质激素类药物。氯替泼诺(loteprednol, 3, 图 1)是用于治疗眼部炎症的药物, 在血浆中氯替泼诺会转变为一种无活性的羧酸代谢产物, 这样可明显降低它的不良反应。环索奈德(ciclesonide, 4)作用于肺部, 可用于治疗哮喘和慢性阻塞性肺病。相比于早期开发的吸入性糖皮质激素, 环索奈德由于能在血浆中快速降解为无活性产物, 因此其具有更高的安全性。氟考丁酯(fluorcortin butyl, 5)为局部皮肤用药, 在血浆中也可迅速降解失活[17]。这些局部给药的酯类化合物在进入血液循环后会迅速通过水解酶水解失活, 从而降低化合物在体内的不良反应。

|
Figure 1 Examples of anti-inflammatory soft drugs |
根据血浆稳定性实验结果, 当化合物结构中存在易水解的基团时, 可以考虑用更加稳定的基团替换不稳定结构, 以提高化合物的血浆稳定性。生物电子等排[4]是改善化合物稳定性的重要结构修饰策略之一。它能以较小的结构变化达到提高化合物血浆稳定性的效果, 同时尽可能保持化合物的药理活性。
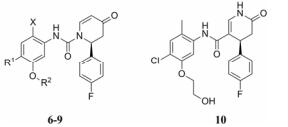
由于酰胺基团相对于相同位置的酯基等易水解基团更稳定, 因此酰胺基团常常被用来替代一些易水解基团。化合物6是由罗氏制药公司开发的一类结构新颖的P2X7受体拮抗剂(表 2)[21], P2X7受体可能在炎症、免疫、神经性疾病和肌肉骨骼疾病等方面发挥重要作用[22]。由于化合物9在人血浆中不稳定, 其在血浆中孵育4 h就有50%的原型药物被降解。因此, 研发人员将化合物结构中的脲基替换为酰胺基团得到化合物10, 从而大大提高了化合物的血浆稳定性, 同时其活性也得到了较好的保持(IC50 = 23 nmol·L-1)。
| Table 2 Plasma stabilities of P2X7 receptor antagonists |
酯基是一类在药物结构中的常见基团, 但有时药物中的酯基在体内易被酯酶水解, 从而导致化合物失去活性。对于血浆水解作用, 酰胺相对于酯更加稳定。研究表明:蛋白激酶B (PKB)的过度激活与肿瘤的发生发展密切相关[23, 24]。化合物11(图 2)是由罗氏制药公司开发的一种蛋白激酶B抑制剂[IC50 (PKB-α) = 5 nmol·L-1][25], 其结构中的酯基在血浆中极其不稳定, 其在小鼠血浆中的半衰期小于1 min。若用酰胺基团替换酯基得到化合物12, 其血浆半衰期可延长至69 h, 并且其对PKB-α的抑制活性也得到了保持[IC50 (PKB-α) = 4 nmol·L-1]。若将酯基替换为醚或者胺(化合物13和14), 尽管化合物的血浆稳定性大大提高(t1/2 = 29~161 h), 但是其抑制活性明显下降。

|
Figure 2 Plasma stabilities of PKB inhibitors |
盐酸普鲁卡因(procaine, 15)是一类短效酯类局麻药(图 3), 其药效维持时间约为30~60 min。造成局部麻醉作用持续时间短的主要原因之一是由于其在体内易水解失效。盐酸普鲁卡因在体内的代谢过程主要是被血浆中的假性胆碱酯酶催化水解。为了延长其麻醉作用时间, 研究人员随后开发出了盐酸利多卡因(lidocaine, 16)。盐酸利多卡因与普鲁卡因结构的主要区别就在于其用酰胺键替换了普鲁卡因结构中的酯键, 酰胺较酯的血浆稳定性更佳。另外, 在利多卡因酰胺键邻位引入两个甲基, 增加了空间位阻作用, 使得利多卡因在体内的水解速度明显减慢, 同时其麻醉作用比普鲁卡因强2~9倍[26]。另外, 仅从体内稳定性角度而言, 将普鲁卡因结构中的酯基替换为酰胺得到抗心律失常药普鲁卡因胺(procainamide, 17), 其在体内的半衰期可提高至3 h左右。

|
Figure 3 Improving the plasma stability of local anesthetics by structural modification |
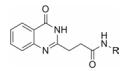
研究发现, 前列腺素D2受体(CRTH2)在炎症和过敏性疾病中发挥重要作用[27]。化合物18是一种由默克公司研发的前列腺素D2受体(CRTH2)拮抗剂(图 4), 该化合物在啮齿类动物和犬体内均展现了较好的药代动力学性质[28]。然而, 在进一步的实验过程中发现化合物18在人血浆中的半衰期(t1/2)约1 h。通过分析代谢产物, 研究人员发现, 该化合物结构中的琥珀酰亚胺环结构易被水解酶水解开环。因此, 他们对化合物18进行结构优化, 将琥珀酰亚胺环替换为内酰胺环得到化合物19。化合物19在人血浆中孵育24 h后仅有不到15%的化合物发生降解代谢, 使其活性得到保持。在药物开发初期阶段评价化合物的血浆稳定性具有重要意义, 它可以避免化合物在进入药物研发后期由于稳定性问题而被迫终止的潜在风险。

|
Figure 4 Improving the plasma stability of CRTH2 receptor antagonists by structural modification |
许多含氮杂环都是酯基和酰胺基的生物电子等排体, 采用含氮杂环替换易水解基团, 可以提高药物的血浆稳定性。头孢噻肟(cefotaximesodium, 20)是一类对革兰阴性菌具有较强活性的抗生素(图 5), 由于其结构中C-3位的酯基在血浆中易被水解, 因此药物化学家将酯基替换为较难水解的四氮唑杂环, 提高了化合物的血浆稳定性, 开发获得新型抗生素如头孢甲肟(cefmenoxime, 21)等[26]。

|
Figure 5 Plasma stability of cephalosporin antibiotics |
化合物22是由葛兰素史克公司开发的人巨细胞病毒(HCMV)蛋白酶抑制剂(表 3), 该化合物在分子水平具有较好的抑制活性和选择性[29], 但是该化合物在人血浆中不稳定(t1/2 = 0.5 h)[8]。血浆稳定性差影响了这类结构的后续研发。通过生物电子等排策略, 将环丙羰基替换为苯环或者芳杂环, 得到的化合物23~25在人血浆中的稳定性均显著提高(t1/2 > 24 h), 其中化合物25对人巨细胞病毒蛋白酶抑制活性也有所提高(IC50 = 0.18 μmol·L-1)。
| Table 3 Improving the plasma stability of HCMV inhibitors by structural modification |
乙酰胆碱(acetylcholine, 26)是体内一种重要的神经递质, 在血液内能迅速被胆碱酯酶破坏(图 6)。为了寻找性质更稳定的拟胆碱药, 人们以乙酰胆碱为先导化合物, 将羰基邻位的甲基替换为氨基, 氨基的供电子效应能够有效降低羰基的亲电性, 另外, 在酯基邻位引入甲基还可以通过增加空间位阻效应, 提高氯贝胆碱(bethanechol chloride, 27)的血浆稳定性[26]。

|
Figure 6 Plasma stability of cholinergic drugs |
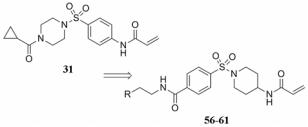
化合物28是由Evotec公司开发的一种组织型转谷氨酰胺酶2 (TG2)抑制剂[30], 研究表明TG2的过度活化与脂泻病和亨廷顿舞蹈病密切相关(表 4)[31, 32]。化合物28对TG2具有较好的抑制活性, 但是其在小鼠血浆中的稳定性较差(t1/2 = 33.5 min)。与其具有类似结构的化合物29和30的血浆稳定性也很差, 其中化合物30结构中由于氨基邻位嘧啶取代的吸电子效应更强, 使其在血浆中被降解的速度更快, 小鼠血浆中的半衰期仅为14.3 min。利用生物电子等排策略, 将结构中的吡啶环或嘧啶环替换为苯环, 降低其取代基的吸电子效应, 使酰胺键不易被水解酶水解, 有助于提高化合物的血浆稳定性。化合物31不仅活性得到保持, 而且在小鼠血浆中的稳定性也得到了明显改善(t1/2 = 209 min)。
| Table 4 Improving the plasma stability of transglutaminase 2 inhibitors by structural modification |
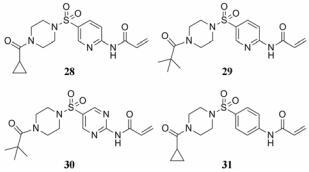
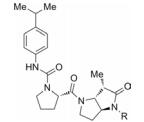
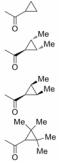
增加易水解基团附近的空间位阻作用, 可降低化合物与水解酶之间的亲和力, 从而提高化合物的血浆稳定性[29]。在前面的例子中(表 3), 化合物22在人血浆中不稳定[29]。研究人员在化合物22的环丙烷部分引入两个甲基取代, 以增加酰胺键附近的空间位阻, 分别得到两个不同构型的化合物32和33, 它们在人血浆中的稳定性均有显著提高, 并且化合物33的酶抑制活性也得到保持(表 5)。在环丙烷环上继续引入两个甲基增加位阻, 尽管化合物34的血浆稳定性增加了, 但是化合物的抑制活性大大下降。因此, 通过增加空间位阻提高化合物的血浆稳定性需要精细修饰, 才能获得较优的改造结果。同时, 从这个例子也可以看出, 手性对化合物的血浆稳定性和药理活性也有重要影响, 这也是在进行结构改造的过程中需要考虑的重要因素。
| Table 5 Improving the plasma stability of HCMV inhibitors by structural modification |
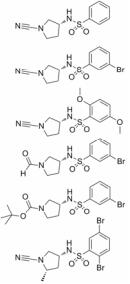
研究表明, 组织蛋白酶C (Cathepsin C)在败血症和炎症相关疾病中具有重要作用[33, 34]。化合物35是由葛兰素史克公司开发的一种组织蛋白酶C抑制剂[35], 由于化合物35在大鼠血浆中的稳定性不佳(表 6), 对其苯环进行衍生化得到化合物36和37, 它们对组织蛋白酶C的抑制活性得到提高, 但是其血浆稳定性并未得到改善。研究人员认为可能是结构中的氰基部分被血浆中水解酶降解, 因此他们将氰基替换为醛基或者叔丁氧羰基得到化合物38和39, 尽管化合物在血浆中很稳定, 但是化合物对组织蛋白酶C的抑制活性基本丧失。鉴于氰基对活性的重要影响, 研究人员考虑通过降低氰基的亲电性同时增加邻位的空间位阻来稳定氰基。在吡咯环C-5位引入甲基得到化合物40, 其在血浆中的稳定性明显提高, 同时化合物对组织蛋白酶C的抑制活性也进一步提高。
| Table 6 Improving the plasma stability of cannabinoid receptor agonists |
化合物41是一种蛋白激酶激活受体1 (PAR1)抑制剂(表 7), 具有抗血小板活化功能[36]。化合物41具有较好的PAR1抑制活性和良好的理化性质, 但是其在大鼠血浆中的稳定性较差。首先, 研究人员采用生物电子等排策略, 将结构中的一个酰胺基替换为苄胺基, 得到化合物42, 但其血浆稳定性并未得到提高。然而, 将另一个酰胺基用烷基胺替代, 得到化合物43, 化合物43在保持活性的同时, 其在人和大鼠的血浆中均能稳定存在(孵育5 h后分别有99%和97%的原型化合物剩余)。通过在酰胺键邻位引入位阻基团得到化合物44和45。尽管化合物44在人和鼠的血浆中足够稳定(孵育5 h后均有99%的原型化合物剩余), 但是其活性大大降低(在5 μmol·L-1浓度时抑制率仅为21%); 而化合物45不仅对PAR1抑制活性得到保持, 同时其在大鼠血浆中的稳定性也得到了明显改善(表 7)。
| Table 7 Plasma stability of a series of PAR1 inhibitors |

成环修饰是药物结构改造中的重要策略[37], 通过限制化合物结构中某些化学键和基团的自由转动, 使其能以特定构象与靶点作用, 从而提高化合物的药理活性和稳定性。多肽类药物的血浆稳定性较差。因此, 提高肽类药物在血浆中的稳定性是药物研发过程中不可忽视的环节。对肽类化合物进行成环修饰, 限制其分子构象, 在不影响药理活性的同时, 降低其与体内水解酶的亲和性, 从而提高肽类药物的稳定性。而在利用成环修饰策略的时候, 研究者需要考虑到成环的位置、氨基酸残基的选择以及环大小等因素[38]。
胰高血糖素样肽-1 (GLP-1)静脉给药后, 其在人体内的消除半衰期小于2 min。化合物47是将8位丙氨酸替换为甘氨酸的GLP-1类似物(表 8), 其表现出了较好的结合活性(IC50 = 2.8 nmol·L-1)和功能活性(EC50 = 0.014 nmol·L-1)。若进一步将若干连续的氨基酸成环得到化合物49~51, 这类化合物均保持了很好的药理活性, 同时它们在人血浆中的稳定性也得到明显改善(图 7)[38]。因此, 利用成环修饰也是一种有效的提高化合物稳定性的结构改造策略。
| Table 8 A series of GLP-1 receptor agonists. *CRE: cAMP response element; Luc: Luciferase |

|
Figure 7 The stability of GLP-1 receptor agonists in human plasma |
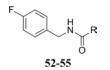
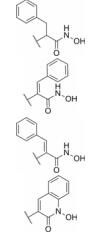
异羟肟酸基团是药物结构中常见的活性片段, 例如伏立诺他(SAHA)结构中就含有该基团。然而, 异羟肟酸基团在血浆中易被水解, 影响了其药代动力学性质和药效作用的发挥。化合物52是一类疟原虫的锌金属蛋白酶抑制剂[39], 其在大鼠血浆中的半衰期只有0.8 h (表 9)。通过在异羟肟酸基团邻位引入双键考察其不同构型对化合物血浆稳定性的影响, 发现化合物(Z)-53的血浆稳定性优于化合物(E)-54, 这可能是由于Z构型中苯环更靠近异羟肟酸基团, 增加了空间位阻作用, 使得水解酶不易接近该部位。进一步结构修饰得到化合物55, 其在血浆中的稳定性进一步提高(t1/2 > 48 h)。尽管文献中未给出这些化合物的药理活性数据, 但是这类改造策略成功提高了化合物的血浆稳定性, 为含有异羟肟酸结构片段化合物的结构改造提供了新的思路。
| Table 9 Rat plasma stabilities of analogues of malonylhy droxamic acid |
前述的化合物31[30]对TG2的IC50为0.11 μmol·L-1, 而其在血浆中的半衰期(t1/2)为3.5 h, 仍然不够长。为了进一步提高化合物31的血浆稳定性, 研究人员利用骨架跃迁的策略, 得到一系列结构新颖的不可逆抑制剂(表 10, 化合物56~61)。这些化合物对TG2的抑制活性均得到保持, 同时它们在人和鼠血浆中的半衰期均大于6 h[40]。这可能是由于脂肪型的酰胺键反应性弱于芳香型的酰胺键, 从而提高了其在血浆中的稳定性。
| Table 10 Improving the plasma stabilities of compounds by scaffold hopping |
端锚聚合酶(tankyrases)属于聚ADP-核糖聚合酶(poly ADP-ribose polymerase, PARP)蛋白家族成员, 它能够促进β-catenin降解, 从而调节Wnt信号通路[41]。抑制tankyrases对于治疗APC (adenoma touspolyposis coli)突变的结直肠癌提供新的研究方向。化合物62是tankyrase-1抑制剂(tankyrase-1, TNKS1, IC50 = 10 nmol·L-1), 但其在大鼠血浆中的半衰期(t1/2)仅为35 min。因此, 为了提高化合物在大鼠血浆中的稳定性, 研究人员对化合物62进行了一系列的结构改造。在酰胺键邻位引入甲基增加位阻效应, 化合物63的血浆半衰期(t1/2 = 43 min)与62相当, 然而其对TNKS1的抑制活性有显著下降(IC50 = 210 nmol·L-1)。在酰胺键的邻位引入甲基得到化合物64, 虽然其在大鼠血浆中的稳定性得到明显改善, 但是其抑制活性下降(IC50 = 1.75 μmol·L-1)。随后研究人员采用骨架跃迁的策略得到环己烷环骨架的化合物65和66, 其中化合物65对TNKS1的抑制活性不仅得到保持, 其在大鼠血浆中的稳定性也大大提高(t1/2 > 1 000 min) (表 11)。
| Table 11 Improving the plasma stability of tankyrases-1 (TNKS1) inhibitors |

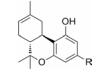
化合物67是一种大麻素受体-1 (cannabinoid receptor-1, CB-1)激动剂[42], 由于该结构中的酯基易水解, 其在大鼠血浆中的半衰期(t1/2)仅为0.7 min。当在酯基邻位引入偕二甲基增加空间位阻时, 化合物68对大鼠的大麻素受体的亲和力提高50倍以上, 同时血浆半衰期(t1/2)也增加了13倍。而引入环丁烷骨架, 化合物69在大鼠血浆中的半衰期进一步提高(t1/2 = 36.3 min), 提升了化合物的血浆稳定性(表 12)。
| Table 12 Improving the plasma stability of cannabinoid receptor-1 (CB1) agonists |

活性化合物的稳定性是影响其成药性的关键因素。化合物的血浆稳定性与肝代谢稳定性有时并不一致, 这是一个经常容易被人们忽视的因素。因此, 在早期先导化合物开发过程中, 有必要开展化合物的血浆稳定性研究, 尽早发现潜在的不稳定结构并对其进行必要的结构修饰, 或者挑选稳定性更佳的先导化合物开展进一步的新药研发。通过将生物电子等排、增加空间位阻、成环修饰以及骨架跃迁这些结构改造策略综合运用, 改善化合物的血浆稳定性, 提高化合物的成药性质, 以期能够为候选药物的开发提供帮助。
| [1] | Kumar GN, Surapaneni S. Role of drug metabolism in drug discovery and development[J]. Med Res Rev, 2001, 21: 397–411. DOI:10.1002/(ISSN)1098-1128 |
| [2] | Jang GR, Harris RZ, Lau DT. Pharmacokinetics and its role in small molecule drug discovery research[J]. Med Res Rev, 2001, 21: 382–396. DOI:10.1002/(ISSN)1098-1128 |
| [3] | Thompson TN. Optimization of metabolic stability as a goal of modern drug design[J]. Med Res Rev, 2001, 21: 412–419. DOI:10.1002/(ISSN)1098-1128 |
| [4] | Wang J, Liu H. Lead compound optimization strategy (1)-changing metabolic pathways and optimizing metabolism stability[J]. Acta Pharm Sin (药学学报), 2013, 48: 1521–1531. |
| [5] | Kerns EH, Di L. Pharmaceutical profiling in drug discovery[J]. Drug Discov Today, 2003, 8: 316–323. DOI:10.1016/S1359-6446(03)02649-7 |
| [6] | Di L, Kerns EH, Hong Y, et al. Development and application of high throughput plasmastability assay for drug discovery[J]. Int J Pharm, 2005, 297: 110–119. DOI:10.1016/j.ijpharm.2005.03.022 |
| [7] | Qin X, Chen A, Lu J, et al. Evaluation of plasma stability and pharmacokinetics of parthenolide in rats with LC-MS/MS analysis[J]. Acta Pharm Sin (药学学报), 2017, 52: 609–614. |
| [8] | Borthwick AD, Exall AM, Haley TM, et al. Pyrrolidine-5, 5-trans-lactams as novel mechanism-based inhibitors of human cytomegalovirus protease. Part 3:potency and plasma stability[J]. Bioorg Med Chem Lett, 2002, 12: 1719–1722. DOI:10.1016/S0960-894X(02)00294-9 |
| [9] | van De Waterbeemd H, Smith DA, Beaumont K, et al. Propertybased design:optimization of drug absorption and pharmacokinetics[J]. J Med Chem, 2001, 44: 1313–1333. DOI:10.1021/jm000407e |
| [10] | Allderdice PW, Gardner HA, Galutira D, et al. The cloned butyrylcholinesterase (BCHE) gene maps to a single chromosome site, 3q26[J]. Genomics, 1991, 11: 452–454. DOI:10.1016/0888-7543(91)90154-7 |
| [11] | Perham RN. The fructose-1, 6-bisphosphate aldolases:same reaction, different enzymes[J]. Biochem Soc Trans, 1990, 18: 185–187. DOI:10.1042/bst0180185 |
| [12] | Jeon YH, Heo YS, Kim CM, et al. Phosphodiesterase:overview of protein structures, potential therapeutic applications and recent progress in drug development[J]. Cell Mol Life Sci, 2005, 62: 1198–1220. DOI:10.1007/s00018-005-4533-5 |
| [13] | Zhou SB, Wang J, Liu H. Lead compound optimization strategy (5)-reducing the hERG cardiac toxicity in drug development[J]. Acta Pharm Sin (药学学报), 2016, 51: 1530–1539. |
| [14] | Cook CS, Karabatsos PJ, Schoenhard GL, et al. Species dependent esterase activities for hydrolysis of an anti-HIV prodrug glycovir and bioavailability of active SC-48334[J]. Pharm Res, 1995, 12: 1158–1164. DOI:10.1023/A:1016259826037 |
| [15] | Yoshigae Y, Imai T, Horita A, et al. Species differences for stereoselective hydrolysis of propranolol prodrugs in plasma and liver[J]. Chirality, 1997, 9: 661–666. DOI:10.1002/(ISSN)1520-636X |
| [16] | Hale JJ, Mills SG, MacCoss M, et al. Phosphorylated morpholineacetal human neurokinin-1 receptor antagonists as water-soluble prodrugs[J]. J Med Chem, 2000, 43: 1234–1241. DOI:10.1021/jm990617v |
| [17] | Ettmayer P, Amidon GL, Clement B, et al. Lessons learned from marketed and investigational prodrugs[J]. J Med Chem, 2004, 47: 2393–2404. DOI:10.1021/jm0303812 |
| [18] | Beaumont K, Webster R, Gardner I, et al. Design of ester prodrugs to enhance oral absorption of poorly permeable compounds:challenges to the discovery scientist[J]. Curr Drug Metab, 2003, 4: 461–485. DOI:10.2174/1389200033489253 |
| [19] | Ji X, Wang J, Zhang L, et al. Application of phosphates and phosphonates prodrugs in drug research and development[J]. Acta Pharm Sin (药学学报), 2013, 48: 621–634. |
| [20] | Sawa M, Tsukamoto T, Kiyoi T, et al. New strategy forantedrug application:development of metalloproteinase inhibitors as antipsoriatic drugs[J]. J Med Chem, 2002, 45: 930–936. DOI:10.1021/jm010349c |
| [21] | Francisco L, Keith AMW, Christine B, et al. Novel series of dihydropyridinone P2X7 receptor antagonists[J]. J Med Chem, 2015, 58: 8413–8426. DOI:10.1021/acs.jmedchem.5b00365 |
| [22] | Bartlett R, Stokes L, Sluyter R. The P2X7 receptor channel:recent developments and the use of P2X7 antagonists in models of disease[J]. Pharmacol Rev, 2014, 66: 638–675. DOI:10.1124/pr.113.008003 |
| [23] | Paweletz CP, Charboneau L, Bichsel VE, et al. Reverse phase protein microarrays which capture disease progression show activation of prosurvival pathways at the cancer invasion front[J]. Oncogene, 2001, 20: 1981–1989. DOI:10.1038/sj.onc.1204265 |
| [24] | Stal O, Perez-Tenorio G, Akerberg L, et al. Akt kinases in breast cancerand the results of adjuvant therapy[J]. Breast Cancer Res, 2003, 5: R37–44. DOI:10.1186/bcr569 |
| [25] | Breitenlechner CB, Wegge T, Berillon L, et al. Structure based optimization of novel azepane derivatives as PKB inhibitors[J]. J Med Chem, 2004, 47: 1375–1390. DOI:10.1021/jm0310479 |
| [26] | Zheng H. Medicinal Chemistry[M]. Ed 6th. Beijing: People's Medical Publishing House Co., LTD, 2007. |
| [27] | Cosmi L, Annunziato F, Galli MIG, et al. CRTH2 is the most reliable marker for the detection of circulating human type 2 Th and type 2 T cytotoxic cells in health and disease[J]. Eur J Immunol, 2000, 30: 2972–2979. DOI:10.1002/(ISSN)1521-4141 |
| [28] | Stefano C, Catherine J, Patrick P, et al. Optimization of the central core of indolinone acetic acid-based CRTH2(DP2) receptor antagonists[J]. ACS Med Chem Lett, 2011, 2: 644–649. DOI:10.1021/ml2001196 |
| [29] | Borthwick AD, Davies DE, Ertl PF, et al. Design and synthesis of pyrrolidine-5, 5'-trans-lactams(5-oxo-hexahydropyrrolo[3, 2-b] pyrroles) as novel mechanism-based inhibitors of human cytomegalovirus protease. 4. Antiviral activity and plasma stability[J]. J Med Chem, 2003, 46: 4428–4449. DOI:10.1021/jm030810w |
| [30] | Prime ME, Andersen OA, Barker JJ, et al. Discovery and structure-activity relationship of potent and selective covalent inhibitors of transglutaminase 2 for huntington's disease[J]. J Med Chem, 2012, 55: 1021–1046. DOI:10.1021/jm201310y |
| [31] | Molberg O, McAdam S, Sollid L. Role of tissue transglutaminasein celiac disease[J]. J Paediatr Gastroenterol Nutr, 2000, 30: 232–240. DOI:10.1097/00005176-200003000-00005 |
| [32] | Siegel M, Khosla C. Transglutaminase 2 inhibitors and their therapeutic role in disease states[J]. Pharmacol Ther, 2007, 115: 232–245. DOI:10.1016/j.pharmthera.2007.05.003 |
| [33] | Mallen-St Clair J, Pham CT, Villalta SA, et al. Mast cell dipeptidyl peptidase Ⅰ mediates survivalfrom sepsis[J]. J Clin Invest, 2004, 113: 628–634. DOI:10.1172/JCI200419062 |
| [34] | Methot N, Rubin J, Guay D, et al. Inhibition of the activation of multiple serine proteases with a cathepsin C inhibitor requires sustained exposure to prevent pro-enzyme processing[J]. J Biol Chem, 2007, 282: 20836–20846. DOI:10.1074/jbc.M702615200 |
| [35] | Laine D, Palovich M, McCleland B, et al. Discovery of novel cyanamide-based inhibitors of cathepsin C[J]. ACS Med Chem Lett, 2011, 2: 142–147. DOI:10.1021/ml100212k |
| [36] | Dockendorff C, Aisiku O, VerPlank L, et al. Discovery of 1, 3-diaminobenzenes as selective inhibitors of platelet activation at the PAR1 receptor[J]. ACS Med Chem Lett, 2012, 3: 232–237. DOI:10.1021/ml2002696 |
| [37] | Koh DW, Dawson TM, Dawson VL. Poly(ADP-ribosyl) ationregulation of life and death in the nervous system[J]. Cell Mol Life Sci, 2005, 62: 760–768. DOI:10.1007/s00018-004-4508-y |
| [38] | Miranda LP, Winters KA, Gegg CV, et al. Design and synthesis of conformationally constrained glucagon-like peptide-1 derivatives with increased plasma stability and prolonged in vivo activity[J]. J Med Chem, 2008, 51: 2758–2765. DOI:10.1021/jm701522b |
| [39] | Flipo M, Charton J, Hocine A, et al. Hydroxamates:relationships between structure and plasma stability[J]. J Med Chem, 2009, 52: 6790–6802. DOI:10.1021/jm900648x |
| [40] | Prime ME, Brookfield FA, Courtney SM, et al. Irreversible 4-aminopiperidine transglutaminase 2 inhibitors for huntington's disease[J]. ACS Med Chem Lett, 2012, 3: 731–735. DOI:10.1021/ml3001352 |
| [41] | Hua Z, Bregman H, Buchanan JL, et al. Development of novel dual binders as potent, selective, and orally bioavailable tankyrase inhibitors[J]. J Med Chem, 2013, 56: 10003–10015. DOI:10.1021/jm401317z |
| [42] | Nikas SP, Sharma R, Paronis CA, et al. Probing the carboxyester side chain in controlled deactivation (-)-Δ8-tetrahydrocannabinols[J]. J Med Chem, 2015, 58: 665–681. DOI:10.1021/jm501165d |