 2018, Vol. 53
2018, Vol. 53



2. 浙江省杭州市第十四中学, 浙江 杭州 310006
2. Hangzhou No. 14 Middle School, Hangzhou 310006, China
泛素化是体内蛋白质降解的重要途径, 蛋白质被泛素化修饰后, 会被蛋白酶体识别并降解。泛素是一种小而高度保守的蛋白质。泛素化酶是执行泛素化的工具, 主要分为3类:泛素激活酶E1、泛素结合酶E2和泛素连接酶E3。泛素化酶发挥作用的过程大致是:泛素会在ATP供能的情况下被E1酶激活, 转移到E2酶上, 紧接着E3酶会通过特异性识别底物, 使之与泛素相连, 最后蛋白酶体识别泛素修饰过的蛋白并将其降解。
除了泛素化酶之外, 细胞中还存在非常重要的去泛素化酶(DUB), 其作用为去除蛋白质上的泛素链, 从而调控蛋白的稳定性和功能。根据DUBs活性位点的不同可以将这些酶分为:泛素特异性蛋白酶(ubiquitin-specific proteases, USPs)、泛素羧基末端水解酶(ubiquitin carboxy-terminal hydrolases, UCHs)、卵巢肿瘤蛋白酶(ovarian-tumor proteases, OTUs)、马查多-约瑟夫病蛋白结构域蛋白酶(Machado-Joseph disease protein domain proteases, MJDs)、JAMM/MPN区域相关金属肽酶(JAMM/MPN domain-associated metallopeptidases, JAMMs)以及单核细胞趋化蛋白诱导蛋白(monocyte chemotactic protein-induced protein, MCPIP)。目前研究最多的是USPs, 其功能较为明确, 且已经发现了不少抑制剂可干预USPs的功能, 成为新型的药物作用靶点[1, 2]。
恶性肿瘤作为人类至今尚未攻克的复杂疾病, 如何高效、精准地杀伤肿瘤一直以来都是相关领域的研究热点。通过分子靶标治疗肿瘤是近年来的新方向, 目前已经报道了多种肿瘤相关靶标和通路, DUB作为其中重要的一类分子靶标, 通过调控机体的蛋白降解等过程, 影响肿瘤的发生、发展。
1 DUB相关通路人体中存在着各种各样的信号通路, 它们通过一系列的分子机制来调控机体的生命活动, 在机体中发挥着重要的调节作用。一旦信号通路的调节受到影响, 就会导致机体的病变。DUB通过调控机体的多种蛋白的降解来影响肿瘤的发生和发展, 而DUB发挥作用的重要途径就是通过调节机体的多种信号通路, 进而促使下游的相关蛋白的含量发生变化, 从而促进或者抑制癌症的发生。研究表明, DUB参与调节Wnt/β-catenin信号传导、转化生长因子β (transforming growth factor-β, TGF-β)、蛋白激酶B (protein kinase B, Akt)、核转录因子κB (nuclear factor kappa-light-chain-enhancer of activated B cells, NF-κB)等多种癌症相关通路。下文将以信号通路进行分类, 对肿瘤相关DUB进行综述。
1.1 Wnt/β-catenin信号传导通路Wnt/β-catenin信号传导通路是生物体内与多种生命活动密切相关的一类传导通路, 其失调会导致一系列的人类疾病尤其是癌症的发生。在这个通路中, β-catenin是一个关键的信号分子, 其含量的增加与肿瘤的发生密切相关。通常情况下, 泛素化系统会维持β-catenin在生物体内含量的稳定性, 一旦去泛素化酶表达增加, 就会导致β-catenin的含量增加, 从而导致肿瘤的发生[3]。
1.1.1 UCH37UCH37能够与转录因子7 (tran scription factor 7, Tcf7)特异性结合而使后者去泛素化, 后者在中胚层形成过程中发挥转录催化剂的作用。Tcf7会与Wnt/β-catenin信号传导通路相关基因结合并促进其的表达, 从而使β-catenin的表达量增加, 进而导致细胞增殖异常以及肿瘤的发生[4]。
1.1.2 USP5USP5是细胞中一种转录因子FoxM1的去泛素化酶。后者在细胞中的主要作用是参与Wnt/β-catenin信号传导通路, FoxM1进入细胞核中, 阻止β-catenin的抑制分子ICAT与之结合, 使更多的β-catenin发挥作用。在细胞中, FoxM1的含量通过泛素化进行平衡, 如果USP5的表达量增多, 就会引起FoxM1的含量增加, 从而使β-catenin的含量增加, 导致细胞增殖速度加快, 发生癌变。已有研究证实, 在结肠癌中, FoxM1会影响β-catenin在细胞中的含量[5]。
1.1.3 UCH-L1研究发现, UCH-L1在细胞癌变的过程中发挥着重要的作用。较新的研究显示, UCH-L1在小儿高级胶质瘤中表达水平较高, 经过一系列的验证, 说明了UCH-L1与小儿高级胶质瘤密切相关, 并且经过实验验证得知: UCH-L1的异常表达促进肿瘤的形成与Wnt/β-catenin信号传导通路有关[6]。
1.1.4 其他USP7调节Wnt/β-catenin信号传导通路, 前者的异常激活会导致人类各种癌症特别是结肠直肠癌的发生, USP7表达量过高, 会增加癌症发生的风险[3]; USP14通过影响Wnt/β-catenin信号传导通路的活性来影响细胞的增殖、转移等过程, USP14的过表达与肝癌密切相关[7]。同样, USP4也是通过调节Wnt/β-catenin信号传导通路的活性从而引发癌症, 在结肠癌细胞中证实, USP4在癌症细胞中的表达量高于正常细胞, 同时, 随着USP4的过表达或者敲除, β-catenin的量也随之增加或者减少。这说明: USP4是β-catenin的一种去泛素化酶, 参与正向调节Wnt/β-catenin信号传导通路。USP4的过表达与癌症的发生密切相关, 是一个潜在的抗癌靶点[8]。多数的DUB都会促进肿瘤的发生, 但少数的DUB起着截然相反的作用。肿瘤抑制因子cylindromatosis (CYLD)就是其中之一, 它能够在肿瘤细胞中表达, 发挥抑制肿瘤细胞增殖扩散的作用。已有研究证实: CYLD在多重骨髓瘤(multiple myeloma, MM)中表达缺失, CYLD通过作用于底物Dvl来发挥抑制多重骨髓瘤生长的作用[9, 10]。
1.2 TGF-β通路TGF-β通路在调节细胞增殖、凋亡、分化和迁移中起关键性的作用。在自然状态下, 组织细胞产生的TGF-β均为静止状态[11]。研究表明: TGF-β通路可以作为癌症治疗的靶点。与正常的组织细胞相比, 肿瘤细胞中TGF-β表达量更高。DUB与TGF-β通路息息相关, 大多数的DUB通过减少相关分子的降解, 从而维持了TGF-β的高浓度, 导致肿瘤的发生和转移[12, 13]。
1.2.1 USP26/USP15Smad特异性E3泛素连接酶2 (SMAD-specific E3 ubiquitin protein ligase 2, SMURF2)是细胞中的一种E3泛素化连接酶, 靶向TGF-β受体(TGF-β receptor, TβR), Smad7在整个过程中发挥脚手架的作用, 使SMURF2发挥泛素化的作用。USP26是Smad7的去泛素化酶, 如果细胞中USP26的含量正常, 则会调控Smad7的表达, 从而使SMURF2发挥作用, 使下游的TβR泛素化。如果USP26表达过少, 则TβR的量过多, 就会导致肿瘤的发生。已经证实, USP26在1 400余例肿瘤细胞系中均存在表达缺失的现象[14]。相反的, USP15是TβR的去泛素化酶, 与Smad7和SMURF2的作用恰好相反, 过量表达USP15可以使下游TβR被泛素化降解的部分变少, 使得整个通路被过度激活。实验证实: USP15的过度表达导致TGF-β通路的过度激活与成胶质细胞瘤的形成密切相关[12]。另有研究表明, USP15同时靶向TGF-β受体以及SMURF2, 使得SMURF2在接触反应中失活, 二者共同的作用使得TGF-β受体含量上升, 与肿瘤的形成密切相关[15]。
1.2.2 USP11USP11是细胞中间变性淋巴瘤激酶(anaplasticlymphoma kinase 5, ALK5)的去泛素化酶, 后者是一种TβR。Smad7在整个过程中发挥脚手架的作用, 招募E3泛素化连接酶, 从而使整个通路被抑制, 而USP11起到的作用与Smad7相反。USP11的过度表达会导致TGF-β通路的过度激活, 实验验证: USP11的敲除会抑制细胞从上皮组织到间叶组织的转移。因此, USP11的过度表达与肿瘤息息相关[13]。
1.2.3 CYLDCYLD抑制肿瘤的发生发展与Wnt/β-catenin信号传导通路有着重要的关联, 同时与TGF-β通路也有着紧密的联系。CYLD也参与Smad7的表达。在远端转移的口腔鳞状细胞癌中, CYLD和Smad7的表达均有所下降。经研究表明: CYLD通过调控Smad7的量来控制下游的TGF-β通路, 而Smad7不会反过来调控CYLD的表达。另有研究证实: CYLD是Smad的一种去泛素化酶。而Smad7是TGF-β通路的一种抑制分子, 从而抑制癌症的发生发展[16]。因此, CYLD可以抑制口腔鳞状细胞癌的转移, 是治疗口腔鳞状细胞癌的一个靶点[17]。
1.3 Akt通路Akt是一种丝氨酸/苏氨酸特异性蛋白激酶, 是很多生长因子(如胰岛素)重要的细胞内信号转导因子。其参与调节细胞的增殖、代谢、转录、迁移和凋亡。Akt通路在生物体内对于细胞的存活和增殖起促进作用, 因此在癌细胞中表达量会增加。泛素化可以调节这一通路的表达量。大多情况下, 当DUB表达量过高时, 就会引起Akt的表达量增加, 进而引发肿瘤的发生等[18, 19]。
1.3.1 USP4USP4不仅对Wnt/β-catenin信号传导通路进行调节, 同时也在Akt通路中发挥着促肿瘤作用。USP4是细胞中肝再生磷酸酶-3 (phosphatase of regenerating liver-3, PRL-3)的去泛素化酶, 后者在细胞中起到酪氨酸磷酸化酶的作用, 与癌细胞的增殖、转移等密切相关, 其在癌细胞中表达量高于正常细胞中。PRL-3在细胞中参与调节磷脂酰肌醇3激酶(phosphatidylinositol 3 kinase, PI3K)/Akt、上皮细胞钙粘蛋白(E-cadherin)、信号传导及转录激活因子(signal transducers and activators of transcription, STAT)、p53、转录因子Snail等信号通路及分子表达。实验验证得到: USP4调控PRL-3介导的PI3K/Akt、E-cadherin等过程。对于PI3K/Akt通路, USP4的表达增加, 会使下游PRL-3的泛素化过程减少, 使PRL-3的含量增多, 从而激活PI3K/Akt通路。如果PI3K/Akt通路被过度激活, 则会引起肿瘤等疾病的发生[20]。
1.3.2 USP22USP22发挥致癌基因的作用是通过过度激活Akt/糖原合成酶3 (glycogen synthase kinase-3, GSK-3)/细胞周期蛋白(cyclin)信号通路实现的。在鼻咽癌细胞实验中发现:该细胞中USP22的表达量明显提高, 影响了细胞生存能力和细胞周期等因素, 从而导致了细胞的癌变。USP22与鼻咽癌的发生密切相关[21]。同时, USP22也影响PI3K/Akt通路的活性。在骨肉瘤细胞中, 其过度表达导致PI3K/Akt通路的活性增加而引发癌症[22]。另有研究表明, USP22表达程度过高, 会引起下游的沉默信息调节因子1 (sirtuin type 1, SIRT1)表达增加, USP22和SIRT1的含量升高都会引起Akt通路被过度激活, 而经研究表明, Akt的过度激活会使肿瘤干细胞产生更强的抵抗药物的作用, 这一作用是肝癌难以治愈的重要原因。因此, USP22的过度表达与癌症的发生有着紧密的联系[23]。
1.3.3 UCH-L1UCH-L1与癌症的发生发展息息相关, 对Wnt/β-catenin信号传导通路有着重要的调控作用。研究证实, UCH-L1在人类多种癌细胞中都存在过表达的现象。它可以提高细胞的生存、迁移和增殖能力。研究表明, UCH-L1促进癌细胞的生长与其提高Akt通路的表达有关[24]。另有研究表明, UCH-L1是通过调控哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin, mTOR)的活性来调控Akt通路的活性。mTOR复合物1 (mTORC1)可以抑制mRNA翻译, 而mTOR复合物2 (mTORC2)则起到促进细胞增殖等作用。二者之间的平衡靠的是UCH-L1抑制mTORC1来调控。如果UCH-L1表达过多, 则mTORC1表达过少, mTORC2表达过多。二者量的失衡会导致癌症的发生[25]。
1.3.4 USP12在细胞中, USP12与USP1相关因子1 (Usp1-associated factor 1, Uaf-1)和WD重复蛋白(WD-repeat protein, WDR20)一起, 是Akt的磷酸化酶丝氨酸/苏氨酸蛋白磷酸酶(pleckstrin homology domain leucine-rich repeat protein phosphatase, PHLPP)和PHLPP类似物(PHLPP-like, PHLPPL)的去泛素化酶, 后者可以将磷酸化的Akt (pAkt)转化为非磷酸化的Akt。在前列腺细胞中, 雄性激素受体(androgen receptor, AR)起着接收雄性激素信号、调控细胞生命活动的作用。pAkt可以将活性AR转化为非活性AR。USP12表达增加, 会使PHLPP和PHLPPL含量增加, 导致pAkt变少, Akt变多, 活性AR变多。同时USP12也是AR的去泛素化酶, 从而使活性AR的量进一步增加。pAkt与下游多种信号因子的激活有关, 而活性AR的过表达则会导致细胞生长等生命活动变快。因此, USP12的这两个功能发挥的作用是相反的, 实验证实, 对于pAkt的作用要强于对AR的作用。因此, USP12是一种抑癌基因[18]。在癌症中, 由于USP12的含量下降, PHLPP家族的PHLPP1的表达量也会因此下调。有研究表明, WD重复蛋白WDR48会与USP12形成复合物, 正向调节PHLPP1的含量。因此, WDR48·USP12可以作为肿瘤细胞生存的一种抑制剂[26]。
1.3.5 Ataxin-3 (ATXN3)ATXN3能够与细胞中的一种组蛋白乙酰化酶环磷腺苷效应元件结合蛋白(cAMP-response element binding protein, CREB)结合并抑制其发挥作用。后者与生物体内Akt通路的抑制基因同源性磷酸酶-张力蛋白基因(phosphatase and tensin homolog, PTEN)的启动子活性密切相关。如果ATXN3过表达, 则CREB含量就会下降, PTEN的含量也会随之下降, 则Akt通路就会被过度激活。因此, 抑制ATXN3的过量表达是治疗癌症的一个现实策略[27]。
1.3.6 USP14USP14可以通过调控Wnt/β-catenin信号传导通路来介导癌症的发生。但在Akt通路中, USP14与之前提及的DUB不太相同, USP14并不是控制Akt通路的活性, 相反, USP14的活性受到Akt的调节, 被磷酸化之后而激活。激活后的USP14会减缓多种蛋白如mTOR等的降解速率, 促使其蛋白降解受阻, 蛋白水平累积[19]。在肿瘤细胞中, 由于Akt通路的抑制基因PTEN受到抑制, Akt通路被过度激活, 继而激活USP14, 导致蛋白酶体调节蛋白降解的速率变慢, 这与癌症的发生密切相关。
1.4 NF-κB通路NF-κB通路是参与炎症等机体反应的转录因子家族, 它的激活会导致多种基因的上调, 并促进许多类型的细胞(包括癌细胞)的存活。目前NF-κB通路的异常激活已在一些肿瘤中被报道。DUB在调控NF-κB通路中起到了重要的作用, 其中一部分DUB起着正向调节作用, 而另一部分则通过与前者不同的机制从而起到了抑制该通路的作用[28, 29]。
1.4.1 OTUB1OTUB1是细胞凋亡抑制剂(the cellular inhibitor of apoptosis, c-IAP)的去泛素化酶, 通过移除K48连接的泛素链从而起到去泛素化的作用。c-IAP会调节肿瘤坏死因子受体(tumour necrosis factor receptor, TNFR)复合物的组装, 后者介导NF-κB激活与表达, 如果c-IAP表达缺失, 那么NF-κB通路的表达会显著下调。而NF-κB通路的激活与癌症的发生有着重要的关系, 因此, OTUB1和c-IAP在未来可能会成为一个新兴治疗靶点[30]。
1.4.2 USP2aUSP2a是细胞中TNFR1和TNFR2的去泛素化酶, 后两者与细胞凋亡密切相关。通过抑制肿瘤坏死因子(tumour necrosis factor, TNF)的上游通路NF-κB通路的活性, USP2a可以提高TNF介导的细胞凋亡。在HeLa等细胞中, USP2a表达量的下调可以保护细胞免受TNF介导的细胞凋亡[31]。
1.4.3 OTULIN/CYLDCYLD在多种信号通路中都发挥着重要的抗肿瘤作用, 而在NF-κB通路中, CYLD则与OTULIN一起, 作为细胞中线性泛素链组装复合物(linear ubiquitin chain assembly complex, LUBAC)的去泛素化酶。后者的泛素化在NF-κB通路中发挥相应的作用, OTULIN/CYLD主要通过与LUBAC的HOIP PUB结构域结合从而使其去泛素化, 这样就达到了抑制NF-κB通路的作用, 从而发挥抗肿瘤作用[28]。另有研究证实, CYLD与癌症的发生也有密切的联系, CYLD在乳腺癌中表达量较低。CYLD抑制NF-κB受体活化剂配体(receptor activator of NF-κB ligand, RANKL)介导的NF-κB通路的活性, 抑制肿瘤的发生[32]。
1.4.4 A20细胞中的NF-κB通路需要很多的泛素化及去泛素化过程才能够得以激活, A20与CYLD一样, 是一种去泛素化酶, 可以阻止NF-κB通路上游的泛素化过程, 从而达到抑制NF-κB通路的作用[29]。
2 DUB抑制剂目前DUB的抑制剂主要以小分子抑制剂为主, 但尚未有抑制剂进入临床阶段研究。下面列举近几年报道的DUB小分子抑制剂(表 1)[33-41]。
| Table 1 The structures of the deubiquitinating enzyme (DUB) inhibitors in recent years |
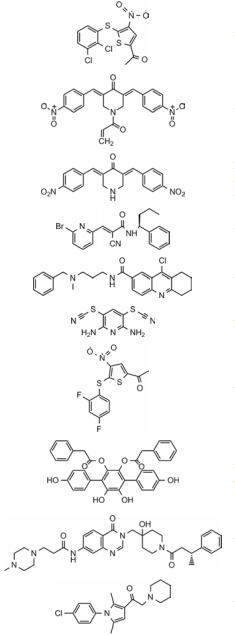
P5091是USP7的一种小分子抑制剂, 前者的过度激活与结直肠癌[3]、慢性淋巴白血病[42]都有一定的关系。
2.2 b-AP15b-AP15是USP14的一种抑制剂, 前者的过度激活会导致白血病细胞的凋亡受到抑制[43], 与食道鳞状细胞癌的发生也密切相关[44]。而通过剂量控制的方式给予b-AP15, 可以增加白血病细胞的凋亡。
2.3 RA-9研究表明, RA-9对于抑制与蛋白酶体相关的DUB的功能有重要的作用。体内治疗发现, RA-9可以延缓肿瘤的生长, 从而起到治疗卵巢癌的作用[35]。另有研究表明, 在乳腺癌细胞中, RA-9可以激活肿瘤细胞的自噬, 从而起到治疗的作用[45]。
2.4 WP1130WP1130是一种部分选择性抑制剂, 它可以抑制USP9X的作用, 后者的过度表达会导致食管鳞状细胞癌、骨髓细胞白血病等肿瘤患者状况变差。WP1130通过抑制USP9X的功能, 使得乳腺癌细胞对于顺铂的敏感度增加, 与后者合用会使治疗效果更好[46]。另有研究显示, WP1130在胰腺导管腺癌中也发挥着很好的抑癌作用, 它会诱导癌细胞死亡, 从而起到治疗的作用[47]。
2.5 HBX 19818USP7是目前肿瘤治疗的一个重要的靶向分子, 研究发现USP7的特异性抑制剂HBX 19818可以选择性地降低USP7的活性, 从而起到治疗癌症的作用[37]。
2.6 PR-619PR-619是一种非特异性的DUB抑制剂, 可以作用于多种DUB[38]。
2.7 P22077P22077是USP7的一种特异性抑制剂[38]。Ki-67抗原是评估非小细胞肺癌细胞增殖的最可信的免疫组织化学标记, 有研究证实: USP7是通过使Ki-67去泛素化, 导致Ki-67水平较高, 从而促进细胞增殖。而P22077通过抑制USP7的作用, 从而起到治疗非小细胞肺癌的效果[48]。研究证实, 在多种肿瘤细胞中, USP7的抑制剂P5091和P22077通过增强细胞内氧化应激反应和内质网应激反应, 介导细胞内活性氧的量增加, 从而引起肿瘤细胞的凋亡[49]。
2.8 Vialinin AVialinin A是USP5及其他一些DUB的抑制剂, USP5已被报道为一种肿瘤治疗靶点。Vialinin A会使NF-κB的抑制分子抑制因子-κB (inhibitor-κB, IκB)的泛素化程度降低, 因此会导致NF-κB通路表达下降, 从而起到治疗肿瘤的作用[39]。
2.9 XL188XL188是USP7的一种选择性抑制剂, 通过与USP7的S4-S5口袋特异性结合来发挥选择性的抑制作用。研究证实, XL188可以提高细胞中的抑癌蛋白P53和P21的含量, 从而抑制肿瘤的发生发展[40]。
2.10 IU1-47IU1-47是USP14的一种特异性抑制剂。研究证实, USP14是通过抑制蛋白酶体的作用来发挥特定的功能的。而IU1-47可以抑制USP14的功能, 使一些蛋白酶体底物的含量下降, 如tau蛋白。通过抑制USP14, IU1-47可以降低细胞中tau蛋白的表达量, 后者的表达与肿瘤的发生发展关系密切。因此, IU1-47可以起到抑制肿瘤的作用[41]。
3 总结与展望泛素化与去泛素化在机体各个方面的平衡与稳定上起到了重要的作用, 目前已经成为癌症治疗与研究的一个重要方向。DUB通过多种信号通路(Wnt/ β-catenin信号传导通路、TGF-β通路、Akt通路、NF-κB通路等)调节肿瘤的生长及癌细胞的扩散(图 1):大多数的DUB促进肿瘤的生长, 而少部分的作用则恰恰相反。目前并未有上市的DUB抑制剂药物。但是在未来, 通过更多的去泛素化酶及相应抑制剂的发现, 通过靶向药物治疗肿瘤有望成为现实。

|
Figure 1 The roles of some DUBs mentioned in this paper in cancer therapy. The red arrows mean the specified DUBs are upregulated in cancer cells while the black ones indicate the opposite. Akt: Protein kinase B; NF-κB: Nuclear factor kappa-light-chain-enhancer of activated B cells; TGF-β: Transforming growth factor-β; USP: Ubiquitin-specific protease; UCH: Ubiquitin carboxy-terminal hydrolase; ATXN3: Ataxin-3; CYLD: Cylin dromatosis |
| [1] | D'Arcy P, Wang X, Linder S. Deubiquitinase inhibition as a cancer therapeutic strategy[J]. Pharmacol Ther, 2015, 147: 32–54. DOI:10.1016/j.pharmthera.2014.11.002 |
| [2] | Wei RB, Liu XD, Yu WX, et al. Deubiquitinases in cancer[J]. Oncotarget, 2015, 6: 12872–12889. |
| [3] | An T, Gong YX, Li X, et al. USP7 inhibitor P5091 inhibits Wnt signaling and colorectal tumor growth[J]. Biochem Pharmacol, 2017, 131: 29–39. DOI:10.1016/j.bcp.2017.02.011 |
| [4] | Han W, Lee H, Han JK. Ubiquitin C-terminal hydrolase37 regulates Tcf7 DNA binding for the activation of Wnt signalling[J]. Sci Rep, 2017, 7: 42590. DOI:10.1038/srep42590 |
| [5] | Chen YH, Li Y, Xue JF, et al. Wnt-induced deubiquitination FoxM1 ensures nucleus β-catenin transactivation[J]. EMBO J, 2016, 35: 668–684. DOI:10.15252/embj.201592810 |
| [6] | Sanchez-Diaz PC, Chang JC, Moses ES, et al. Ubiquitin carboxyl-terminal esterase L1(UCHL1) is associated with stem-like cancer cell functions in pediatric high-grade glioma[J]. PLoS One, 2017, 12: e0176879. DOI:10.1371/journal.pone.0176879 |
| [7] | Huang G, Li LM, Zhou W. USP14 activation promotes tumor progression in hepatocellular carcinoma[J]. Oncol Rep, 2015, 34: 2917–2924. DOI:10.3892/or.2015.4296 |
| [8] | Yun SI, Kim HH, Yoon JH, et al. Ubiquitin specific protease 4 positively regulates the WNT/beta-catenin signaling in colorectal cancer[J]. Mol Oncol, 2015, 9: 1834–1851. DOI:10.1016/j.molonc.2015.06.006 |
| [9] | Van Andel H, Kocemba KA, De Haan-Kramer A, et al. Loss of CYLD expression unleashes Wnt signaling in multiple myeloma and is associated with aggressive disease[J]. Oncogene, 2017, 36: 2105–2115. DOI:10.1038/onc.2016.368 |
| [10] | Tauriello DVF, Haegebarth A, Kuper I, et al. Loss of the tumor suppressor CYLD enhances Wnt/beta-catenin signaling through K63-linked ubiquitination of Dvl[J]. Mol Cell, 2010, 37: 607–619. DOI:10.1016/j.molcel.2010.01.035 |
| [11] | Ma Y, Liu H, Zhang H, et al. The TGF-β signaling pathway induced EMT in breast cancer[J]. Acta Pharm Sin (药学学报), 2015, 50: 385–392. |
| [12] | Eichhorn PJA, Rodon L, Gonzalez-Junca A, et al. USP15 stabilizes TGF-beta receptor Ⅰ and promotes oncogenesis through the activation of TGF-beta signaling in glioblastoma[J]. Nat Med, 2012, 18: 429–435. DOI:10.1038/nm.2619 |
| [13] | Al-Salihi MA, Herhaus L, Macartney T, et al. USP11 augments TGF beta signalling by deubiquitylating ALK5[J]. Open Biol, 2012, 2: 120063. DOI:10.1098/rsob.120063 |
| [14] | Lui SKL, Lyengar PV, Jaynes P, et al. USP26 regulates TGFbeta signaling by deubiquitinating and stabilizing SMAD7[J]. EMBO Rep, 2017, 18: 797–808. DOI:10.15252/embr.201643270 |
| [15] | Iyengar PV, Jaynes P, Rodon L, et al. USP15 regulates SMURF2 kinetics through C-lobe mediated deubiquitination[J]. Sci Rep, 2015, 5: 14733. DOI:10.1038/srep14733 |
| [16] | Zhao YG, Thornton AM, Kinney MC, et al. The deubiquitinase CYLD targets Smad7 protein to regulate transforming growth factor beta (TGF-beta) signaling and the development of regulatory T cells[J]. J Biol Chem, 2011, 286: 40520–40530. DOI:10.1074/jbc.M111.292961 |
| [17] | Ge WL, Xu JF, Hu J. Regulation of oral squamous cell carcinoma proliferation through crosstalk between SMAD7 and CYLD[J]. Cell Physiol Biochem, 2016, 38: 1209–1217. DOI:10.1159/000443069 |
| [18] | Mcclurg UL, Summerscales EE, Harle VJ, et al. Deubiquitinating enzyme Usp12 regulates the interaction between the androgen receptor and the Akt pathway[J]. Oncotarget, 2014, 5: 7081–7092. |
| [19] | Xu D, Shan B, Lee BH, et al. Phosphorylation and activation of ubiquitin-specific protease-14 by Akt regulates the ubiquitinproteasome system[J]. Elife, 2015, 4: e10510. |
| [20] | Xing C, Lu XX, Guo PD, et al. Ubiquitin-specific protease 4-mediated deubiquitination and stabilization of PRL-3 is required for potentiating colorectal oncogenesis[J]. Cancer Res, 2016, 76: 83–95. |
| [21] | Zhuang YJ, Liao ZW, Yu HW, et al. shRNA-mediated silencing of the ubiquitin-specific protease 22 gene restrained cell progression and affected the Akt pathway in nasopharyngeal carcinoma[J]. Cancer Biol Ther, 2015, 16: 88–96. DOI:10.4161/15384047.2014.987029 |
| [22] | Zhang DF, Jiang F, Wang X, et al. Downregulation of ubiquitinspecific protease 22 Inhibits proliferation, invasion, and epithelialmesenchymal transition in osteosarcoma cells[J]. Oncol Res, 2017, 25: 743–751. DOI:10.3727/096504016X14772395226335 |
| [23] | Ling SB, Li J, Shan QN, et al. USP22 mediates the multidrug resistance of hepatocellular carcinoma via the SIRT1/AKT/MRP1 signaling pathway[J]. Mol Oncol, 2017, 11: 682–695. DOI:10.1002/mol2.2017.11.issue-6 |
| [24] | Frisan T, Coppotelli G, Dryselius R, et al. Ubiquitin Cterminal hydrolase-L1 interacts with adhesion complexes and promotes cell migration, survival, and anchorage independent growth[J]. Faseb J, 2017, 26: 5060–5070. |
| [25] | Hussain S, Feldman AL, Das C, et al. Ubiquitin hydrolase UCH-L1 destabilizes mTOR complex 1 by antagonizing DDB1-CUL4-mediated ubiquitination of raptor[J]. Mol Cell Biol, 2013, 33: 1188–1197. DOI:10.1128/MCB.01389-12 |
| [26] | Gangula NR, Maddika S. WD repeat protein WDR48 in complex with deubiquitinase USP12 suppresses Akt-dependent cell survival signaling by stabilizing PH domain leucine-rich repeat protein phosphatase 1(PHLPP1)[J]. J Biol Chem, 2013, 288: 34545–34554. DOI:10.1074/jbc.M113.503383 |
| [27] | Sacco JJ, Yau TY, Darling S, et al. The deubiquitylase Ataxin-3 restricts PTEN transcription in lung cancer cells[J]. Oncogene, 2014, 33: 4265–4272. DOI:10.1038/onc.2013.512 |
| [28] | Takiuchi T, Nakagawa T, Tamiya H, et al. Suppression of LUBAC-mediated linear ubiquitination by a specific interaction between LUBAC and the deubiquitinases CYLD and OTULIN[J]. Genes Cells, 2014, 19: 254–272. DOI:10.1111/gtc.12128 |
| [29] | Wang Y, Park NY, Jang YM, et al. Vitamin E gammatocotrienol inhibits cytokine-stimulated NF-kappa B activation by induction of anti-inflammatory A20 via stress adaptive response due to modulation of sphingolipids[J]. J Immunol, 2015, 195: 126–133. DOI:10.4049/jimmunol.1403149 |
| [30] | Goncharov T, Niessen K, De Almagro MC, et al. OTUB1 modulates c-IAP1 stability to regulate signalling pathways[J]. EMBO J, 2013, 32: 1103–1114. DOI:10.1038/emboj.2013.62 |
| [31] | Mahul-Mellier AL, Pazarentzos E, Datler C, et al. Deubiquitinating protease USP2a targets RIP1 and TRAF2 to mediate cell death by TNF[J]. Cell Death Differ, 2012, 19: 891–899. DOI:10.1038/cdd.2011.185 |
| [32] | Hayashi M, Jono H, Shinriki S, et al. Clinical significance of CYLD downregulation in breast cancer[J]. Breast Cancer Res Treat, 2014, 143: 447–457. DOI:10.1007/s10549-013-2824-3 |
| [33] | Chauhan D, Tian Z, Nicholson B, et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance[J]. Cancer Cell, 2012, 22: 345–358. DOI:10.1016/j.ccr.2012.08.007 |
| [34] | D'Arcyl P, Brnjic S, Olofsson MH, et al. Inhibition of proteasome deubiquitinating activity as a novel cancer therapy[J]. Nat Med, 2011, 17: 1636–1640. DOI:10.1038/nm.2536 |
| [35] | Coughlin K, Anchoori R, Lizuka Y, et al. Small-molecule RA-9 inhibits proteasome-associated DUBs and ovarian cancer in vitro and in vivo via exacerbating unfolded protein responses[J]. Clin Cancer Res, 2014, 20: 3174–3186. DOI:10.1158/1078-0432.CCR-13-2658 |
| [36] | Leestemaker Y, De Jong A, Ovaa H. Activity-Based Proteomics:Methods and Protocols[M]. New York: New York Humana Press, 2017: 113-130. |
| [37] | Reverdy C, Conrath S, Lopez R, et al. Discovery of specific inhibitors of human USP7/HAUSP deubiquitinating enzyme[J]. Chem Biol, 2012, 19: 467–477. DOI:10.1016/j.chembiol.2012.02.007 |
| [38] | Altun M, Kramer HB, Willems LI, et al. Activity-based chemical proteomics accelerates inhibitor development for deubiquitylating enzymes[J]. Chem Biol, 2011, 18: 1401–1412. DOI:10.1016/j.chembiol.2011.08.018 |
| [39] | Okada K, Ye YQ, Taniguchi K, et al. Vialinin A is a ubiquitin-specific peptidase inhibitor[J]. Bioorg Med Chem Lett, 2013, 23: 4328–4331. DOI:10.1016/j.bmcl.2013.05.093 |
| [40] | Lamberto I, Liu XX, Seo HS, et al. Structure-guided development of a potent and selective non-covalent active-site inhibitor of USP7[J]. Cell Chem Biol, 2017, 24: 1490–1500. DOI:10.1016/j.chembiol.2017.09.003 |
| [41] | Boselli M, Lee BH, Robert J, et al. An inhibitor of the proteasomal deubiquitinating enzyme USP14 induces tau elimination in cultured neurons[J]. J Biol Chem, 2017, 292: 19209–19225. DOI:10.1074/jbc.M117.815126 |
| [42] | Carra G, Panuzzo C, Torti D, et al. Therapeutic inhibition of USP7-PTEN network in chronic lymphocytic leukemia:a strategy to overcome TP53 mutated/deleted clones[J]. Oncotarget, 2017, 8: 35508–35522. |
| [43] | Song C, Ma R, Yang X, et al. The deubiquitinating enzyme USP14 regulates leukemic chemotherapy drugs-induced cell apoptosis by suppressing ubiquitination of aurora kinase B[J]. Cell Physiol Biochem, 2017, 42: 965–973. DOI:10.1159/000478679 |
| [44] | Zhang BH, Li MQ, Huang PZ, et al. Overexpression of ubiquitin specific peptidase 14 predicts unfavorable prognosis in esophageal squamous cell carcinoma[J]. Thoracic Cancer, 2017, 8: 344–349. DOI:10.1111/tca.2017.8.issue-4 |
| [45] | Vogel RI, Coughlin K, Scotti A, et al. Simultaneous inhibition of deubiquitinating enzymes (DUBs) and autophagy synergistically kills breast cancer cells[J]. Oncotarget, 2015, 6: 4159–4170. |
| [46] | Fu PF, Du FY, Liu Y, et al. WP1130 increases cisplatin sensitivity through inhibition of USP9X in estrogen receptornegative breast cancer cells[J]. Am J Transl Res, 2017, 9: 1783–1791. |
| [47] | Cox JL, Wilder PJ, Wuebben EL, et al. Context-dependent function of the deubiquitinating enzyme USP9X in pancreatic ductal adenocarcinoma[J]. Cancer Biol Ther, 2014, 15: 1042–1052. DOI:10.4161/cbt.29182 |
| [48] | Zhang C, Lu J, Zhang QW, et al. USP7 promotes cell proliferation through the stabilization of Ki-67 protein in non-small cell lung cancer cells[J]. Int J Biochem Cell Biol, 2016, 79: 209–221. DOI:10.1016/j.biocel.2016.08.025 |
| [49] | Lee G, Oh TI, Urn KB, et al. Small-molecule inhibitors of USP7 induce apoptosis through oxidative and endoplasmic reticulum stress in cancer cells[J]. Biochem Biophys Res Commun, 2016, 470: 181–186. DOI:10.1016/j.bbrc.2016.01.021 |