 2018, Vol. 53
2018, Vol. 53

新药发现与研究实例简析
新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
首创性药物经历的艰辛过程, 在于对靶标的可药性和对先导物结构全然未知, 在反复探究的互动中相互验证而相得益彰。杜邦公司的研发者“捡起”武田的苗头化合物, 通过分子模拟比对苗头与配体结构的异同, 对分子结构的各个位置作“地毯式”的变换与考察, 通过逐步构效关系分析, 做阶段性的理性推断, 成为下一轮设计的起点。对比氯沙坦(56)与苗头物(2)的结构, 分子骨架发生了巨大的变化, 外周的药效团虽依稀存在, 但体内外活性已有显著提升。氯沙坦的研制彰显出先导物优化中丰富的药物化学内涵。
(编者按)
肾素-血管紧张素系统(renin-angiotensin system, RAS)是20世纪60年代就已证实在维持心血管的正常发育、电解质和体液平衡以及调节血压等方面起主要作用。肝脏中产生的血管紧张素原是种糖蛋白, 在蛋白水解酶肾素(renin)的催化作用下, 裂解成十肽血管紧张素Ⅰ (Ang Ⅰ), 后者经血管紧张素转化酶(ACE)的作用, 裂解成八肽血管紧张素Ⅱ (Ang Ⅱ), Ang Ⅱ具有收缩外周血管升高血压作用。
在RAS系统中至少有这三个作用环节可作为降低高血压治疗心血管疾病的靶标, 即肾素抑制剂、ACE抑制剂和血管紧张素Ⅱ受体阻断剂。图 1列出了由血管紧张素原生成Ang Ⅱ的生化过程和药物干预的环节(靶标)。

|
图 1 肾素-血管紧张素系统和药物干预的环节 |
RAS系统早在19世纪就已经知晓, 但并没有循此路径研发降压新药。在20世纪70年代以前, 降压药主要为β-阻断剂、α-阻断剂、利尿剂和钙通道拮抗剂等, 由于这些药物都有不同程度的不良反应, 人们致力于研究以RAS为作用环节的药物。
20世纪60年代阐明了ACE的功能, 成为率先研究的作用靶标, 目标是可口服的ACE抑制剂, 于1981年上市了降压药卡托普利(captopril), 是在ACE的功能和活性化合物的互动中, 成功上市的首创药物, 这也确证了ACE的可药性(druggability)。后续跟进了更多的“普利”类降压药。
普利类虽然没有已往降压药的不良反应, 却有引起干咳和血管性水肿等副作用, 这是因为ACE类似于激肽酶Ⅱ, 可水解血浆中的激肽和P物质, ACE抑制剂导致激肽和P物质在血浆中水平增高, 引起上述不良反应。因而促使人们研究肾素-血管紧张素系统的另一个靶标—血管紧张素Ⅱ受体拮抗剂, 旨在阻断Ang Ⅱ的功能, 不干扰ACE酶的功能。
血管紧张素Ⅱ受体有两种亚型: AT1和AT2, Ang Ⅱ受体拮抗剂的降压作用主要是抑制AT1受体。
1.3 血管紧张素Ⅱ的结构特征血管紧张素Ⅱ为八肽(1), 通过对各个氨基酸的变换考察对AT1受体的激动/拮抗作用, 提示结构中的3个芳香氨基酸: Tyr4、His6和Phe8是必须的残基, His6对于受体-配体的分子识别至关重要, Tyr4和Phe8是必要的激动性基团, 其余5个氨基酸起辅助或肽链构象的支撑作用。Tyr4-Ile5形成弯曲构象, Pro7的特殊结构构成了Pro7-Phe8键转折, 这样使得Tyr4、His6和Phe8侧链的芳环簇集在一起, 这已由二维核磁共振研究所揭示(Matsoukas JM, Bigam G, Zhou N, et al. 1H-NMR studies of [Sar1] angiotensin Ⅱconformation by nuclear overhause effect spectroscopy in the rotating frame (ROESY): clustering of the aromatic rings in dimethylsulfoxide. Peptides, 1990, 11: 359-366)。
Tyr4、His6和Phe8残基之间还存在内在的协同性作用:由于残基之间的靠近效应, Phe8的羧基负离子经过His6咪唑环的质子传递, 促进了Tyr4酚羟基失去质子, 形成稳定的酚负氧离子, 在与AT1结合和引发效应中起重要作用。该三元体的电子传递与活化与丝氨酸蛋白酶的三元体Asp-His-Ser的电子传递有相似之处, 如图 2所示。此外, Ile5占据的疏水空间也是拮抗剂结合的位点。

|
图 2 AT1中Phe-His-Tyr三元体电子转移示意图 |
武田药厂在研究利尿降压药中发现化合物2 (CV-2198)既有利尿作用也有降压活性, 并且发现在呈现降压作用的剂量下, 并不产生利尿作用, 证明了是由于选择性地抑制Ang Ⅱ受体所致(IC50 = 40 μmol·L-1)。武田经优化得到的化合物3进行了临床研究, 虽然有降压和利尿作用, 但降血压作用微弱, 从而放弃了该项目的研究, 未料到这个放弃却成就了杜邦(后并入默克)公司。
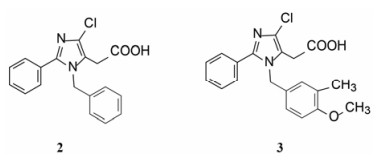
|
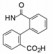
杜邦公司对于开发口服的AT1受体拮抗剂有浓厚兴趣, 苗头化合物载于武田专利(Furukawa Y, Kishimoto S, Nishikawa K. US Patent, 4355040, 1982), 其中化合物4~6对AT1有选择性弱抑制作用。遂以化合物6为模版, 用分子模拟方法比较了6与Ang Ⅱ的结构特征, 发现二者某些基团或片段在结构或性质上具有相似与对应性, 在此基础上进行了如下的分析和推理: ① AT1受体结合的配体分子为八肽, 6显示较弱的拮抗作用可能是分子过于短小, “遮盖”不住受体活性部位。加大6的分子尺寸应有利于提高活性。② Ang Ⅱ的C-端羧基与AT1的互补性正电基团发生静电引力, 例如6的羧基是必要的功能基, 因为羧基被酯化或酰胺化活性显著降低。推测乙酸基相当于C端的Phe8的羧基, 以负离子形式与受体的正电荷结合。③分子模拟将化合物6的羧基与Ang Ⅱ的C-羧基尽可能叠合, 此时咪唑环的氮原子相当于Ang Ⅱ的组氨酸的咪唑片段。④苄基相当于Ang Ⅱ的N-端疏水性残基位置, 正丁基则对应于Val3的疏水性异丙基侧链。

为了将苗头化合物过渡到先导物(hit-to-lead), 以化合物6为起始物, 对咪唑环上各个位置的取代基进行了变换, 进行广泛的构效关系研究, 分别或(尤其是)同时变换苯环的取代、羧甲基、氯原子和正丁基等, 探索各部位对活性的影响。在对构效关系未知的情况下同时变换两个或三个位置的基团, 而不是逐个单一位点的变换, 可以节省化合物的合成数量, 避免发生化合物“大面积”无效的囧地, 获得事半功倍的效果。深入地考察特定位置的构效关系则在先导物的优化阶段(Duncia JV, Chiu AT, Carini DJ, et al. The discovery of potent nonpeptide angiotensin I1 receptor antagonists: a new class of potent antihypertensives. J Med Chem, 1990, 33: 1312-1329)。
3.1 以苯基取代为主的变换首先考察苯环的4位基团变化对活性的影响, 代表性的化合物及其活性列于表 1中。
| 表 1 代表性的化合物7~19的结构和抑制AT1受体的活性 |


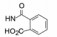

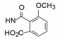
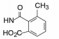
分析表 1化合物的构效关系, 可概括如下: ①苯环的4位连接羧基(化合物7)活性比无取代基的提高一个数量级。有利于提高活性的原因, 推测是模拟了底物Tyr4的羟基或Asp1的羧基, 提供了负电荷与受体的正电荷结合, 增强了对受体的亲和力。若4位用氨基、硝基或甲氧基取代(化合物11~13)失去了活性, 佐证了羧基的重要性。②咪唑环上的羧甲基被酯化或换成羟甲基或甲氧甲基不影响活性, 例如化合物8的活性与7相同。③苯环的4-氨基被邻苯二甲酰单酰化, 化合物14 IC50 = 0.14 μmol·L-1, 推测增加分子的长度, 增加了与受体结合的机会, 并且仍存在对结合有利的羧基。苯基酰胺的邻位若再被取代, 例如甲氧基、甲基等(化合物17和18)活性更高, 提示2, 6位的取代基限制酰胺键的转动, 使分子构象更有利于同受体结合。④以联苯的结构引出羧基的化合物19也有较高的活性, 可理解为增加了分子长度、疏水性和存在位置适宜的羧基, 这些都有利于同受体结合。图 3是对上述邻苯二甲酰胺类化合物构效关系的示意图。

|
图 3 邻苯二甲酰胺类化合物的构效关系 |
羧基是必须的药效团特征。为了调整化合物的亲脂性-亲水性的平衡, 以及提高分子的代谢稳定性, 用三氟甲磺酰胺基或四唑基替换羧基, 亲脂性较强的三氟甲磺酰胺基因三氟甲磺酰基的拉电子作用使氮上的氢原子呈酸性(氮负离子被稳定); 代谢稳定的四唑基的环上唯一氢原子的离解常数pKa = 4 (负离子离域于4个氮原子上而稳定)。合成的代表性化合物列于表 2中。
| 表 2 酸性基团及其位置变换对活性的影响 |
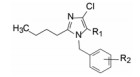

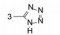

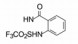
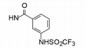


表中化合物的结构与活性的关系提示如下: ①三氟甲磺酰基的强拉电子效应使氨基上的氢呈易离解的酸性氢, 化合物20的活性与相应的羧酸化合物7相当; 同样, 25的三氟甲磺酰胺基也是化合物15的羧基电子等排体, 活性基本相同。②三氟乙酰胺基(化合物21)或甲磺酰胺基化合物24的氢的酸性弱, 活性比相应的化合物15活性显著低。③化合物的三氟甲磺酰胺基若移至间位或对位例如26和27, 活性降低, 提示邻位取代的必要性。④羧基被四唑基置换如化合物22的活性与7相同, 但四唑环改变位置如化合物23的活性则减弱。
3.3 连接基胺酰片段改换成酰胺基-NHCO-→ -CONH-与咪唑环相连的苄基经胺酰键引出的2-羧基苯基(或2-四唑基苯基)显示有较强的抑制AT1受体的活性, 考察该胺酰片段(-NHCO-)的连接方向对活性的影响, 合成了有代表性的酰胺(-CONH-)连接的化合物29~31, 列于表 3中。分析结构与活性表明, 酰胺连接基构成的化合物低于相应的胺酰基化合物, 例如化合物29的活性低于15大约20多倍; 30的活性比相应的18活性低15倍。因而, 两个苯环之间应以胺酰基的方向连接。在末端苯环的羧基邻位引入基团如化合物30的活性显著提高, 与前述的三氟磺酰胺系列引入邻位基团的增效效果相同。将化合物29的羧基改换成四唑基, 活性也有较大的提升, 是因为增加了亲脂性有利于结合(Duncia JV, Chiu AT, Carini DJ, et al. The discovery of potent nonpeptide angiotensin I1 receptor antagonists: a new class of potent antihypertensives. J Med Chem, 1990, 33: 1312-1329)。
| 表 3 胺酰连接基(逆向酰胺)的结构与活性 |

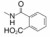



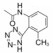
优化至此, 已确定了咪唑环上优化的基团为2-丁基、4-氯和5-羧甲基(或羟甲基、甲氧甲基), 末端苯环存在邻位的酸性基团。然而尚未对两个苯环之间的连接基作系统的考察, 即连接基的长度和走向对活性的影响。为此设计的新化合物是变换苯环之间连接基的长度和原子组成, 表 4列出了化合物的结构和活性。
| 表 4 变换苯基之间的连接基的化合物结构和活性 |

连接基的变换引起活性的变化可归纳如下: ①以化合物14为基准, 胺酰基片段呈共轭平面结构, 连接的两个苯环为反式构象, 变换为单原子连接的羰基, 化合物32和33活性保持不变, 二苯醚(39)、二苯硫醚(40)和联苯化合物41的活性与14相近, 提示单原子或单键相连的化合物的活性与胺酰基的双原子的连接基本不变。②以氧亚甲基-OCH2-连接的化合物例如34~36, 活性低于单原子(或单键)连接的化合物, 可能是3个连续的柔性键使得活性构象难以固定。③反式乙烯基连接形成的菧化合物活性显著降低, 推测分子的形状和基团的空间走向发生了改变。脲基连接的化合物38活性也较低。
研究至此, 对合成的高活性化合物进行了动物实验, 用肾型高血压大鼠静脉注射10~45 mg·kg-1, 一些化合物可降低血压超过30 mmHg。然而经口给药除化合物除45外都没有降压作用。45灌胃肾型高血压大鼠11 mg·kg-1, 可降低血压30 mmHg (Carini DJ, Duncia JV, Johnson AL et al. Nonpeptide angiotensin I1 receptor antagonists: N-[(benzyloxy) benzyl] imidazoles and related compounds as potent antihypertensives. J Med Chem, 1990, 33: 1330-1336; Carini DJ, Duncia JV, Aldrich PE, et al. Nonpeptide angiotensin Ⅱ receptor antagonists: the discovery of a series of N-(biphenylylmethyl)imidazoles as potent, orally active antihypertensives. J Med Chem, 1991, 34: 2525-2547)。
4 里程碑式的先导物——联苯化合物 4.1 口服有效的2'-羧酸联苯化合物上述用单原子连接两个苯环的化合物体外显示活性高, 体内注射肾动脉结扎大鼠有明显降血压作用, 但经口给药无效, 说明这些化合物胃肠道未能吸收。然而由单键连接的联苯化合物如41、44和45, 体外和大鼠注射给药都显示活性, 但灌胃给模型大鼠只有45有效, 如表 5所示。
| 表 5 联苯化合物的羧基位置对体内外活性的影响。*ED30表示降低大鼠血压30 mmHg的灌胃剂量 |
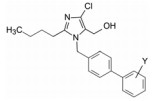
化合物41和45体外抑制AT1的活性相当, 但灌胃给药只有45显示体内有效。曾解释为邻位羧基处于强疏水性的联苯“角落”中, 羧基的极性被掩蔽, 因而有助于过膜吸收, 而3-羧基未被掩蔽。然而, 测定41和45的分配系数, log P值分别是1.17和1.38, 相差无几, 说明不是溶解和过膜性的问题。
4.2 2'-羧基的置换端基苯环的邻位具有酸性基团如羧基对于抑制AT1受体是必要的。用羧基的电子等排体酰胺基团替换羧基, 如表 6所示的构效关系, 若R2是没有酸性的CONH2 (47)基本没有活性, 酰羟胺(48~50)略显酸性, 则稍有活性; 化合物51和52的氨基同时与酰化和磺酰化连接, 双方拉电子提高了酸性强度, 抑制活性与2'-羧基相当; 而将酰胺的连接方向变换为胺酰基如三氟甲磺酰胺54的活性最强, 而55的R1为甲氧甲基, 活性稍弱。然而这些化合物的体内活性都不如羧基化合物45。
| 表 6 羧基的酰胺类电子等排体置换对活性的影响。*此处的IC50定义是, 化合物抑制50%[3H]血管紧张素Ⅱ (2 nmol·L-1)与大鼠肾上腺皮质微粒体结合的浓度。**除ED30表示降低大鼠血压30 mmHg外, 其余数值均为降低血压15 mmHg。 |
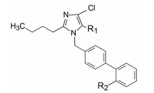
化合物45的羧基pKa为5.0, 用其他酸性基团替换, 如-CONHOH、-CONHSO2Ph、NHCOCF3或NHSO2CF3, 它们的pKa为4.5~10.5, 虽然体外对AT1有抑制活性, 但灌胃大鼠均无降压作用。
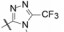
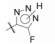
4.3 2'-酸性基团被四(三)唑环取代将R2酸性基团换作四唑基, 化合物56的体内外活性显著提高, 不仅强于相应的羧基化合物45, 而且口服灌胃的活性强于注射途径, 说明四唑基有利于提高化合物的生物利用度, 可能是由于离解后的负电荷均匀地分散在环上, 弥散的负电荷更有利于过膜和结合的缘故。然而换成三唑的化合物58~61, 即使引入拉电子基团例如氰基、羧酯基或三氟甲基, 对离体受体或体内活性都显著降低, 可能是环上的取代基的位阻效应, 不利于同受体结合。表 7列出了四唑和三唑环取代的结构与活性。
| 表 7 四唑和三唑环取代的化合物结构与活性。*此处的IC50定义是, 化合物抑制50%[3H]血管紧张素Ⅱ (2 nmol·L-1)与大鼠肾上腺皮质微粒体结合的浓度。**除ED30表示降低大鼠血压30 mmHg外, 其余数值均为降低血压15 mmHg |



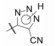

为了探讨四唑基与苯环的相对位置对活性的影响, 在两环之间加入单原子或两原子间隔基, 分别为62和63 (表 8), 活性下降1个数量级, 提示该酸性基团不能拉长; 将四唑环移至苯环的3-或4位(化合物65和66)活性也下降, 表明酸性基团在2位对呈现活性的不可动摇性。化合物67~69是在末端2'-四唑苯环上引入第二个取代基, 活性也显著降低, 提示苯环与受体结合的腔穴空间有限, 似乎不能容忍多余的基团存在。
| 表 8 四唑基于末端苯环的连接状态与活性的相关性。*此处的IC50定义是, 化合物抑制50%[3H]血管紧张素Ⅱ (2 nmol·L-1)与大鼠肾上腺皮质微粒体结合的浓度。**除ED30表示降低大鼠血压30 mmHg外, 其余数值均为降低血压15 mmHg |
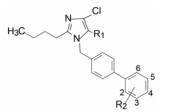
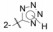
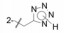

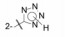

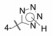
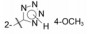

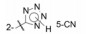
考察咪唑环上4位取代基对活性的影响, Cl、Br、I、H或CF3等取代基的体外活性大都相近, 但灌胃给药降低大鼠血压的活性仍以化合物56最佳(表 9)。
| 表 9 咪唑环的4位取代基对活性的影响。*此处的IC50定义是, 化合物抑制50%[3H]血管紧张素Ⅱ (2 nmol·L-1)与大鼠肾上腺皮质微粒体结合的浓度。**除ED30表示降低大鼠血压30 mmHg外, 其余数值均为降低血压15 mmHg |

以联苯基的2'位固定为羧基, 咪唑环4位固定为氯、5位为羟甲基作为模板化合物, 合成了咪唑2位为乙基、正丙基、正丁基、正戊基、正己基和反式1-丁烯基等不同的化合物, 体外具有相近的活性, 体内以正丁基和反式1-丁烯基活性最强, 提示4个碳原子的长度最佳。
固定2-正丁基-4-氯取代, 联苯基的2'位为羧基或四唑基, 变换咪唑5位的极性基团, 例如羟甲基、氨甲基、各种酰胺甲基、羧基、甲氧羰基、甲酰基或氨酰基等片段, 体内外实验表明活性最强的取代基是羟甲基和羧基(Carini DJ, Duncia JV, Aldrich PE, et al. Nonpeptide angiotensin I1 receptor antagonists: the discovery of a series of N-(biphenylylmethy1) imidazoles as potent, orally active antihypertensives. J Med Chem, 1991, 34: 2525-2547)。
5 候选化合物的确定和氯沙坦的上市以上叙述的有代表性化合物中, 56显示了体外和体内的强效抑制受体和降压活性, 特别是经胃肠道吸收, 对肾型高血压大鼠呈现显著降压作用, 加之由于适宜的药代动力学性质, 研发的杜邦公司(后并入默克)将其确定为候选化合物, 命名为氯沙坦(losartan), 进入临床研究, 于1995年由默克公司开发上市, 成为第一个口服降压的血管紧张素Ⅱ受体拮抗剂。
6 氯沙坦的代谢活化氯沙坦在体内被CYP2C9和3A4氧化代谢, 羟甲基转变成羧基化合物(74, EXP-3174), 口服剂量大约14%转变成74, 其抑制AT1的活性高于氯沙坦40倍, 而且半衰期也长于氯沙坦。这个代谢活化过程启示了后续的沙坦类药物的分子设计。
|
根据氯沙坦的药效团与AngⅡ重要功能基分布的相似性, 以及研发历程中揭示的构效关系, 推测出氯沙坦与AT1受体的结合方式, 映射出氯沙坦呈现药理作用的结构特征, 也为后续研究的沙坦药物提供了设计依据。图 4是氯沙坦与AT1受体结合模式的示意图。

|
图 4 氯沙坦与AT1受体结合模式的示意图 |
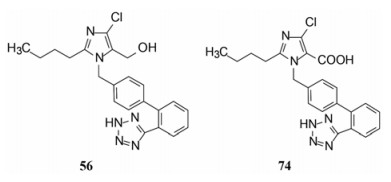
氯沙坦作为首创药物, 前无借鉴, 研发历程长。后续的缬沙坦(75, valsartan)作为模拟创新药物, 采用了药效团和骨架迁越的策略。保持了联苯四唑的骨架和亲脂性的正丁基, 但剖裂了咪唑环成简化的酰胺, 酰基的平面性模拟了吡唑环的亚胺片段, 并巧妙的使用缬氨酸, 既构建了羧基, 又使异丙基代替亲脂性的氯原子。缬沙坦由BMS研发于1995年上市(Carini DJ, Duncia JV, Aldrich PE et al. Nonpeptide angiotensin Ⅱ receptor antagonists: the discovery of a series of N-(biphenylmethyl)-imidazoles as potent, orally active antihypertensives. J Med Chem, 1991, 34: 2525-2547)。
厄贝沙坦(76, irbesartan)保持氯沙坦的大部分结构片段, 但用二氢吡唑酮替换羟甲基吡唑, 氯沙坦的氯原子用亲脂性的4位的螺戊基代替。厄贝沙坦由赛诺菲-安万特研发, 于1998年上市(Bernhart CA, Perreaut PM, Ferrari BP, et al. A new series of imidazolones: highly specific and potent nonpeptide at1 angiotensin ii receptor antagonists. J Med Chem, 1993, 36: 22, 3371-3380)。
|
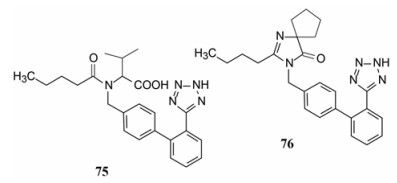
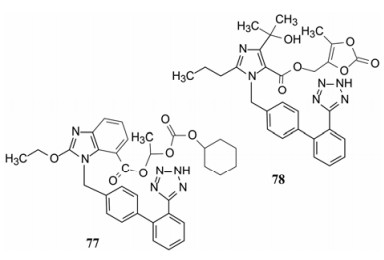
这是两个前药型的AT1受体拮抗剂。坎地沙坦酯(77, candesartan cilrxetil)的骨架结构是苯并咪唑甲酸连接四氮唑联苯甲基, 预构的羧基不利于过膜吸收, 制备成活泼酯以掩蔽羧基的负电荷。在胃肠道吸收后, 被酯酶迅速水解, 释放出坎地沙坦。坎地沙坦与AT1受体结合, 复合物的离解速率常数很小, 因而一次给药有长时间作用。坎地沙坦酯由武田药厂研发, 1997年批准上市(Nishikawa K, Naka T, Chatani F, et al. Candesartan cilexetil: a review of its preclinical pharmacology. J Hum Hypertens, 1997, Suppl 2: S9-17)。
|
奥美沙坦酯(78, omlesartan medoxomil)与氯沙坦的骨架结构相同, 正丁基变为正丙基, 氯原子被羟异丙基代替, 羟甲基用羧基替换, 后者酯化成前药提高了口服生物利用度(Brunner HR. The new oral angiotensin Ⅱ antagonist olmesartan medoxomil: a concise overview. J Hum Hypertens, 2002, Suppl 2: S13-16)。