 2017, Vol. 52
2017, Vol. 52

编者按
新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
在现代药物研发史上阿托伐他汀占有重要一页。作为安全有效的降脂、心血管保护药, 曾是多年的销售额全球第一的重磅药物, 年销售过百亿美元。然而阿托伐他汀的研制并非首创, 而是个跟随性的、全球第五个上市的他汀药物, 它以后来居上的姿态雄踞鳌头, 虽然有一定商业运作因素, 但主要是其品质优良。本文简要介绍了阿托伐他汀研发的药物化学过程。
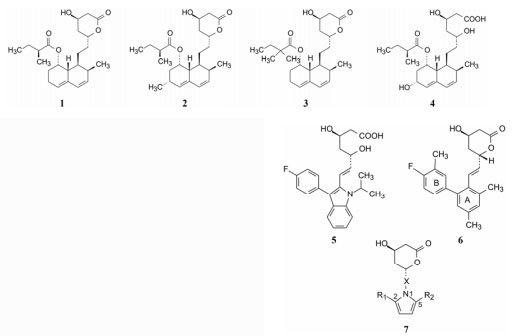
体内低密度脂蛋白(LDL)胆固醇水平提高, 引起动脉硬化和冠心病。人体中胆固醇三分之二是体内合成的, 由乙酰辅酶A经大约30步生化反应合成。其中的限速反应是羟基甲基戊二酸(HMG)在HMG-CoA还原酶催化下, 生成二羟基甲基戊酸, 抑制后者的生成, 切断了胆固醇的生物合成链(Tavormina PA, Gibbs MH, Huff JW. The utilization of β-hydroxy-β-methyl-δ-valerolactone in cholesterol biosynthesis. J Am Chem Soc, 1956, 78: 4498-4499)。
1.2 项目启动时的同类研发状态 1.2.1 美伐他汀的发现20世纪70年代日本三共制药的远藤等在橘青霉菌(Penicillium citrium)中发现抗生素美伐他汀(1, mevastatin, 又称compactin), 证明是HMG-CoA还原酶抑制剂, 可降低实验犬和猴的血浆中低密度脂蛋白胆固醇和总胆固醇。但进入临床研究, 因犬的长期毒性实验呈现有致癌作用而终止。这也为后继的洛伐他汀研究投下了阴影(Endo A, Kuroda M, Tsujita Y. ML-236A, ML-236B, and ML-236C, new inhibitors of cholesterogenesis produced by Penicillium citrium. J Antibiot (Tokyo), 1976, 29: 1346-1348)。
1.2.2 洛伐他汀率先上市受远藤工作的启发, 1978年默沙东公司的Alberts从土曲霉菌(Penicillium terreus)分离出另一个抑制HMG-CoA还原酶的天然成分, 即洛伐他汀(2, lovastatin, 原称mevinolin) (Alberts AW, Chen J, Kuron G, et al. Mevinolin: a highly potent competitive inhibitor of hydroxymethylglutaryl-coenzyme A reductase and a cholesterol-lowering agent. Proc Natl Acad Sci U S A, 1980, 77: 3957-3961), 化学结构与美伐他汀极其相似, 只在六氢萘环上多一个甲基。经过曲折的历程(因美伐他汀的致癌而一度停顿), 于1987年上市, 为首创性的降胆固醇药物, 他汀类第一个药物(first in class)。
1.2.3 辛伐他汀和普伐他汀的上市洛伐他汀在专利和市场销售方面, 在三共与默沙东公司间出现的纠纷很快被它们各自研制的半合成的产品上市而消解, 默沙东在美伐他汀的基础上成功地研发出辛伐他汀(3, simvastatin), 于1988年在瑞典上市。三共在美伐他汀的六氢萘环上引入羟基, 称作普伐他汀(4, pravastatin), 是打开的内酯环为二羟基戊酸的结构, 于1989年在日本上市。
1.2.4 氟伐他汀上市山度士公司研发的他汀摒弃了六氢萘母核, 以吲哚为骨架, 支撑二羟基戊酸的药效团, 连接了必要的疏水片段, 成功地上市了第一个合成的药物氟伐他汀(5, fluvastatin), 于1994年在英国上市。
|
1982年启动阿托伐他汀项目时, 上面4个药物仍在研发阶段, 但相继的上市给阿托伐他汀的研发增添了难度和阻力。
1.3 已有合成化合物的信息上节简述的4个他汀药物的上市, 并没有给阿托伐他汀研制者以设计上的启示, 因为项目研发时间颇多交盖, 并且在专利上设置了禁区, 构效关系也不明朗。
启动该项目, 借鉴已有的结构信息是默克公司Willard等合成的有强效抑制HMG-CoA还原酶活性的化合物6 (Stokker GE, Alberts AW, Anderson PS, et al. 3-Hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors. 3.7-(3, 5-Disubstituted [l, l'-biphenyl]-2-yl)-3, 5-dihydroxy-6-heptenoic acids and their lactone derivatives. J Med Chem, 1986, 29: 170-181), 并作了如下的推定: ① 4-(R)-羟基吡喃-2-酮(即羟基六元内酯)是一个重要的药效团特征, 开环形成羟基戊酸占据HMG与酶结合的位置。②结构下半部连接大体积的亲脂性片段。③苯环B相当于美伐他汀的2-丁酸酯部分, 结合于疏水腔中。2-丁酸酯若被水解, 失去亲脂的丁酸基, 活性下降100倍, 提示疏水性B环也是一个药效团特征。④分子模拟表明B环位置相当于辅酶A的结合腔。
1.4 以吡咯为核心的骨架基于上述的设想, 以及为了研制的药物结构具有新颖性, Parke-Davis公司用吡咯环作为核心结构, N1位通过连接基连接羟基内酯片段, C2和C5连接不同的亲脂性基团, 通式7作为初始的先导物设计, 变换与优化这3个位置。
2 结构优化 2.1 活性评价洛伐他汀和辛伐他汀其实是前药, 羟基内酯在体内开环成活化形式(类似于羟基甲基戊二酸)。所以测定受试物活性需将内酯水解成开环结构。用两种体外模型评价。一种是评价化合物对胆固醇合成的抑制作用(CSI), 用大鼠肝匀浆催化14C标记的乙酸转化成胆固醇的反应, 测定转化的速率, 在一定时间内生成的胆固醇越少, 表明化合物的活性越高, 并以受试物抑制胆固醇生成指定量的50%浓度作为活性指标(IC50, CSI)。另一方法是评价对HMG-CoA还原酶的抑制作用(COR), 用部分纯化的微粒体酶催化14C标记的HMG-CoA转化成甲羟戊酸, 评价受试物对转化反应的抑制活性, 用IC50, COR表示。此外, 也用与美伐他汀的IC50比值来表示。实验表明这两种测定方法具有平行相关性。
2.2 优化吡咯环与内酯环的连接基将吡咯的2位(R1)和5位(R2)分别固定为4-氟苯基和甲基或异丙基, 变换吡咯与内酯环间的连接基, 合成的化合物列于表 1。结果表明, 苯环作连接基的化合物活性很低, 两个亚甲基相连的活性(化合物13)高于三碳连接的化合物11, 因而以后优化R1和R2时, 连接基固定为亚乙基。
| 表 1 变换连接基的化合物及其活性。a比值=美伐他汀IC50, CSI/受试物IC50, CSI) × 100, 下同。每次评价的化合物同时测定美伐他汀的活性, 由于活性的变差, 化合物之间的IC50与相对活性不成比例。但比值之间有可比性 |
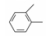
将N1的连接基固定为亚乙基, 5位为甲基, 变换2位的苯环上的取代基, 合成的化合物列于表 2。
| 表 2 2-取代苯基化合物的结构与活性 |
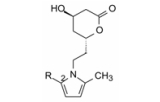
表 2的构效关系提示取代基的变换对活性影响不显著, 其中4-氟苯基化合物13活性最强。
2.4 2位用大体积基团取代的效应进而用芳香或脂肪双环连接于2位, 考察大体积基团对活性的影响, 化合物结构列于表 3。结果表明化合物虽呈现活性, 但不如取代苯基的活性强。
| 表 3 双环取代2位的化合物活性 |
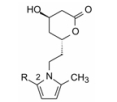


至此, 吡咯与内酯环的连接基以亚乙基占优, 2位优选的片段为4-氟苯基。下一步是固定上述优选的片段, 考察5位取代对活性影响。表 4列出的化合物构效关系提示, 化合物13、30与36的抑制胆固醇合成的活性较强, 而相对活性最强的是30, 判断异丙基为优选的基团。
| 表 4 变换5位取代基的化合物活性 |
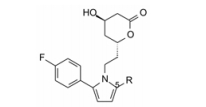
先导化合物的多位点优化往往不是一次完成的, 由于分子内基团间的影响和靶标的构象变化, 需要反复验证各位点的优势片段。表 5列出的化合物是固定连接基为亚乙基、5位为异丙基, 再次变换2位的基团。结果提示仍以4-氟苯基为优化的片段(化合物30)。氟原子在苯环上变换位置(37、38)或二氟(39), 或用甲氧基、甲基、氯原子等取代, 都使活性下降。
| 表 5 变换2位取代基的化合物活性 |

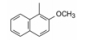
综合上述优化吡咯的1、2和5位取代基结果, 分别是1位连接基为亚乙基, 2位是4-氟苯基, 5位为异丙基, 代表性化合物30是高活性化合物。然而30的活性只是美伐他汀活性的30%, 所以仍需优化(Roth BD, Ortwine DF, Hoefle ML, et al. Inhibitors of cholesterol biosynthesis. 1. trans-6-(2-Pyrrol-l-ylethyl)-4-hydroxypyran-2-ones, a novel series of HMG-CoA reductase inhibitors. 1. Effects of structural modifications at the 2-and 5-positions of the pyrrole nucleus. J Med Chem, 1990, 33: 21-31)。
表 4中化合物36为5-三氟甲基, 活性强于13和30, 推测由于拉电子效应降低了吡咯环的密度而有利于与酶结合。吡咯的3位和4位尚属空位, 未做优化探索。下一步是对这两个位置进行变换。
3 吡咯环的3和4位取代基的优化 3.1 影响电性的基团取代—3, 4-二溴化合物的夭折表 6列出了吡咯环3和4位引入不同取代基的化合物。47是用两个甲基取代, 与30相比, 活性略有提高。甲基是弱推电子基团(以及超共轭效应)与前述拉电子基团有利于活性的效应相悖, 可能是甲基的亲脂性的正贡献抵消并超过了推电性(负贡献)的缘故。化合物48和49分别是二氯和二溴取代物, 活性强于30, 达到美伐他汀活性的79%, 可解释为氯和溴兼有拉电子性(σCl = 0.23; σBr = 0.23)和亲脂性(πCl = 0.71; πBr = 0.86), 对活性都呈正贡献的缘故。由于3, 4-二溴化合物49显示较高活性, 探索了开发前景, 但发现49的大鼠亚急性毒性有毒性反应, 从而终止了对49的研发(Sigler RE, Dominick MA, McGuire EJ. Subaute toxicity of a halogenated pyrrole hydroxymethylglutaryl-coenzyme A reductase inhibitor in Wistar rats. Toxicol Pathol, 1992, 20: 595-602)。化合物50为三氟乙酰基取代, 活性下降, 可能是极性过强不利于结合。两个简单酯基化合物51和52的活性与30相近, 也提示较强的脂溶性可能是提高活性的因素。
| 表 6 变换3和4位取代基的化合物结构与活性。a比值=美伐他汀IC50, COR/受试物IC50, COR) × 100, 下同 |
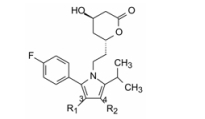
将2位基团固定为4-氟苯基, 5位为异丙基, 表 7列出了在3或4位连接苯环或吡啶环的化合物活性。化合物53与30的活性相似, 而2-吡啶基(54)的活性显著增强, 苯环由3位移至4位, 化合物57的活性有所提高。这些提示引入单个芳香环对活性的影响不显著。
| 表 7 连接芳环的化合物活性 |
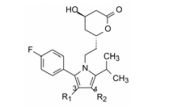
在3和4位连接亲脂性苯环和拉电子基团例如酯基或酰胺基, 合成的有代表性的化合物列于表 8。化合物58和59是局域异构体, 4位的苯基比3位的活性高。从报道的数据看, 研发者更偏重于3位为苯基, 例如苯环上引入4-氰基(化合物60), 活性略有下降, 但4位变成苄酯化合物61, 活性提高。进而变换成4-酰基苯胺, 消旋化合物62的活性可达到美伐他汀活性的80%以上。拆分成光活体62 (4'R)的活性显著提高, 是美伐他汀的5倍, 而4'S活性弱。
| 表 8 3位和4位亲脂性和电性变换的化合物 |

化合物62 (4'R)对HMG-CoA还原酶呈现非常高的活性, 用鼠肝HMG-CoA还原酶评价其活性(IC50 = 0.6 nmol·L-1), 强于同时评价的洛伐他汀(IC50 = 2.7 nmol·L-1)和普伐他汀(IC50=5.5 nmol·L-1)。62 (4'R)的组织分布主要在肝脏, 放射性同位素标记的实验表明, 62 (4'R)在肝脏比在其他组织的分布高30~250倍(肝脏是胆固醇合成的主要器官), 而洛伐他汀没有这种特异性分布。此外, 62 (4'R)还降低大鼠的甘油三酯水平。药代动力学实验表明, 62 (4'R)血浆中半衰期为14 h, 而抑制HMG-CoA还原酶活性的半衰期为20~30 h。口服生物利用度为14%, 抑制HMG-CoA还原酶活性换算为30%, 这些说明62 (4'R)代谢产物仍有活性(代谢活化)。因此62 (4'R)确定为候选化合物, 定名为阿托伐他汀(atorvastatin), 从1982年项目启动到1989年临床前研究结束, 历时7年(Roth BD, Blankley CJ, Chucholowski AW, et al. Inhibitors of cholesterol biosynthesis. 3. Tetrahydro-4-hydroxy-6-[2-(lH-pyrrol-l-yl)ethyl]-2H-pyran-2-one inhibitors of HMG-CoA reductase. 2. Effects of introducing substituents at positions three and four of the pyrrole nucleus. J Med Chem, 1991, 34: 357-366)。
4 阿托伐他汀的批准上市在阿托伐他汀是否进入临床研究的决策上有一个插曲。考虑到有3个他汀药物已经上市, 氟伐他汀即将批准上市, 公司领导层担心上市第五个能否获得足够的市场份额, 拟作出了停止研发的决定(因为根据统计一般认为第四个同类药物的市场份额最多为10%)。项目负责人“据理”陈述阿托伐他汀的优点, 甚至不惜单膝下跪, 恳请临床一试, 最后终于得以进入临床研究。三期临床试验用洛伐他汀、辛伐他汀、普伐他汀和氟伐他汀作对照, 表明在同等剂量下阿托伐他汀降低总胆固醇和甘油三酯等指标都占优胜, 遂于1996年批准上市。
四期临床试验更进一步证实阿托伐他汀是后来居上的降胆固醇药物, 加之成功的商业运作, 使阿托伐他汀连续数年成为全球年销售额第一的重磅药物。阿托伐他汀虽然是第四个跟进药物, 一跃而为同类最佳(best in class)。
5 阿托伐他汀与靶标的结合Istvan和Deisenhofer研究了包括阿托伐他汀的一系列他汀药物与HMG-CoA还原酶复合物的结构生物学(Istvan ES and Deisenhofer J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science, 2001, 292: 1160-1164)。阿托伐他汀的羟甲基戊酸片段的取向与结合方式与其他他汀相同, 都结合于底物羟基甲基戊二酸所处的位置, 羧基与Arg692的胍基形成盐键, 4'-羟基与Asp690形成氢键, 1'-羟基与Lys691、Glu559和Asp767形成氢键网络。图 1是阿托伐他汀与HMG-CoA还原酶复合物晶体衍射图, 点线表示氢键和盐键相互作用, 数字为距离。为了使还原产物甲基二羟基戊酸容易从活性部位释放出去, 并非底物的所有极性基团都参与了结合(Km微摩尔级), 而抑制剂纳摩尔级的Ki值更多是由分子下部的疏水片段的结合所贡献。2位的4-氟苯基(氟原子与4'-羟基形成氢键以稳定构象, 这也是3-F或2-F活性弱的原因)和3位苯环与酶的Leu853、Ala856和Leu857发生疏水结合和范德华作用, 5位异丙基与Leu562有疏水作用。阿托伐他汀的一个重要特征是C4引出的酰胺片段, 酰胺的羰基与Ser565羟基形成氢键(距离为2.7Å), 对Ki值有较大贡献, 使得结合自由能(ΔG)的焓(ΔH)与熵(-TΔS)贡献各占大约50%, 而前四个上市的他汀没有这种结合, 结合的ΔG主要是由熵所驱动(Sarver RW, Bills E, Bolton G, et al. Thermodynamic and structure guided design of statin based inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A reductase. J Med Chem, 2008, 51: 3804-3813)。

|
图 1 阿托伐他汀与HMG-CoA还原酶复合物晶体衍射图 |