 2017, Vol. 52
2017, Vol. 52



2. 中国科学院大学, 北京 100049
2. University of Chinese Academy of Sciences, Beijing 100049, China
DNA缠绕在由组蛋白H2A、H2B、H3和H4各2个分子构成的8聚体上组成核小体, 核小体经过压缩形成染色质, 染色质经过压缩形成染色体。组蛋白H3和H4的氨基末端从核小体突出, 并且其特定位置的氨基酸可被共价修饰, 如磷酸化、乙酰化、泛素化和甲基化等[1]。这些发生于组蛋白上的翻译后修饰可以进一步影响基因的转录。研究表明, 与修饰相关的蛋白主要可分为3类: ① 写入蛋白—可以在组蛋白残基加上特定修饰, 比如组蛋白甲基转移酶、组蛋白乙酰转移酶等; ② 擦除蛋白—可以去除这些修饰, 比如组蛋白去甲基化酶、组蛋白去乙酰化酶等[2]; ③ 读者蛋白—可以识别这些修饰, 比如溴结构域识别蛋白(bromodomains, BRDs)可以识别乙酰化的赖氨酸。
1992年, Tamkun等[3]在果蝇和雌性不育同源蛋白中共同发现了一个结构域并命名其为溴结构域(bromodomains, BRDs)。溴结构域识别乙酰化的赖氨酸从而影响基因转录调控。经序列分析, 在人类基因组的46种不同的蛋白上发现61个溴结构域[4]。按序列同源性可将61个溴结构域分为八大家族, 其中对BET (bromo-and extra-terminal)家族研究最为深入。BET家族包括在组织中广泛表达的BRD2、BRD3、BRD4和主要在睾丸组织中表达的BRDT[5]。BET家族主要功能是招募转录调节复合物到乙酰化的染色质上, 从而控制涉及细胞增殖和细胞周期进展的特定的基因网络。例如BRD4和BRDT可以通过与正转录延伸因子(positive transcriptional elongation factor b, p-TEFb)作用调控转录延长, 导致RNA聚合酶Ⅱ激活引起基因表达[6]。1999年, Dhalluin等[7]第一次用核磁共振(nuclear magnetic resonance, NMR)解析出P300/CBP相关因子PCAF溴结构域的三维结构, 揭示了它具有一个独特的左旋四螺旋结构。随后越来越多的结构生物学研究表明, BRDs在三维结构上均由4个α-螺旋(αZ、αA、αB、αC)组成的一组反平行束[8], 在远离N端和C端的一端αZ和αA螺旋形成疏水性的ZA-loop环, αB和αC螺旋形成疏水性BC-loop环, 它们又形成一个较深的结合乙酰化赖氨酸(KAc)的疏水口袋。溴结构域蛋白的异常与很多疾病密切相关, 比如癌症、炎症、免疫性疾病以及心血管疾病等[9, 10]。如溴结构域蛋白BRD4通过识别组蛋白上乙酰化的赖氨酸从而招募p-TEFb, 进而磷酸化RNA聚合酶Ⅱ, 最终引起下游基因表达, 如癌基因c-Myc基因等。而这些基因的表达异常会导致癌症等疾病。另外一些异常的溴结构域蛋白可能形成致癌突变的融合体, 如TY-82细胞中BRD4基因易位到睾丸核蛋白(NUT)基因座中产生BRD4-NUT融合蛋白, 进而导致c-Myc过表达, 促进NUT中线癌(NMC)的形成[11, 12]。
寻找蛋白—蛋白相互作用的小分子抑制剂被认为是具有挑战性的工作。在2010年报道了两个BET家族选择性抑制剂(+)-JQ1 (1, 图 1)[5]和I-BET762 (2, 图 1)[13], 从而引起人们对溴结构域识别蛋白抑制剂研究的广泛关注。(+)-JQ1被广泛用作化学探针以研究BET蛋白的生理学功能以及它与疾病的关系。I-BET-762是GSK公司通过表型筛选得到的一个BET蛋白抑制剂, 抑制巨噬细胞产生促炎性蛋白质, 并抑制急性炎症, 对其他溴结构域蛋白具有高度选择性, 使得它快速进入临床Ⅱ期试验, 主要用来治疗睾丸中线癌。自从CDK抑制剂dinaciclib被报道也可结合于BRDT的乙酰化赖氨酸结合位点以来[14], 对激酶抑制剂作为溴结构域调节剂的研究吸引越来越多的关注。Sch nbrunnet等[15]通过共晶筛选方法评估了581种不同激酶抑制剂对BET (以BRD4 BD1为代表)的结合潜力。其中PLK1抑制剂BI-2536 (3, 图 1)以及JAK2抑制剂TG101209 (4, 图 1)对BRD4有很好的活性, 文献报道IC50分别是25 nmol·L-1和130 nmol·L-1[16]。这些发现激发人们从激酶抑制剂中合理设计激酶-溴结构域识别蛋白的双重抑制剂或者选择性溴结构域识别蛋白抑制剂。本研究的研究目标是从PLK1-BET溴结构域的双重抑制剂BI-2536来合理设计, 以获得新颖骨架的BRD4抑制剂。

|
Figure 1 Structures of selective BET inhibitors (+)-JQ1 (1), I-BET762 (2), and dual kinase-BET inhibitors BI-2536 (3) and TG101209 (4) |
对化合物3结合BRD4(Ⅰ)(PDB ID: 4OGI)的晶体结构进行分析, 表明3的甲基化酰胺作为ε-N-乙酰化的赖氨酸的模拟基团, 与N140的侧链酰胺形成直接氢键作用, 同时与Y97形成由水介导的氢键作用, 而甲基延伸到由F83、M132和C136形成的疏水口袋中。此外, 乙基突出到小的疏水口袋(V87/L92/L94/Y97) 中, 并且环戊基部分和N-甲基哌啶指向溶剂, 不与附近的残基形成直接作用[17]。所以化合物3对BRD4的高度亲和性主要是由于二氢蝶啶氧与Asn140的相互作用, 而另外与Pro82和ZA通道的水相互作用可以稳定复合物。在PLK1中(PDB ID: 2RKU), 化合物3通过氨基嘧啶部分与铰链区结合, 而二氢蝶啶氧与ATP结合位点的守门残基Leu130形成由水介导的相互作用。所以BI-2536 (3)与BRD4和PLK1的结合模式不同, 为了设计新颖结构的溴结构域识别蛋白BRD4的抑制剂, 可以保留与BRD4形成关键氢键作用的内酰胺结构, 比如用二氢喹喔啉-2(1H)-酮代替BI-2536中的7, 8-二氢蝶啶-6-(5H)-酮。同时也叠合比较了化合物3与BRD4-BD1的共晶结构和本课题组之前报道的一类2-噻唑酮类化合物5 (图 2B)[18]与BRD4-BD1的共晶结构(图 2A), 从图 2A中推测在二氢喹喔啉-2(1H)-酮母核的C6位置增加取代基可能引入基团与WPF亚口袋形成范德华作用。所以接下来的重点就是修饰C6位置的取代基, 希望能找到活性较好的化合物进行后续评价。

|
Figure 2 A : Superimpose crystal structure of BI2536 (3) and 5 bound to BRD4-BD1; B: Chemical structure of 5 |
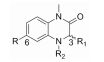
目标化合物的结构经1H NMR、13C NMR和ESI验证, 理化数据见表 1、2, 合成如合成路线1和2所示。在合成路线1中, 商业可得原料4-溴-3-氟-1-硝基苯(30)与D-丙氨酸发生芳香亲核取代(SNAr)反应生成中间体31, 其在连二亚硫酸钠和碳酸钾条件下关环生成中间体32。中间体32与环戊酮发生还原胺化反应得到中间体33, 通过氢化钠使33的酰胺NH去质子化, 随后通过加入碘甲烷引入甲基得到中间体34。中间体34与联硼酸频哪醇酯发生偶联反应生成中间体35, 中间体35与不同取代的溴苯化合物发生Suzuki偶联反应得到目标化合物6 ~25。除了在合成路线1的步骤a中用L-丙氨酸代替D-丙氨酸之外, 其他操作步骤与目标化合物6相似来制备目标化合物26和27。在合成路线2中, 商业可得原料4-溴-3-氟-1-硝基苯(30)与环丙胺发生SNAr反应生成中间体36, 随后用铁粉和氯化铵还原36得到中间体37。中间体37与2-溴丙酰溴发生缩合和分子内亲核取代两步反应得到中间体38, 通过氢化钠使38的酰胺NH去质子化, 随后通过加入碘甲烷引入甲基得到中间体39。中间体39与联硼酸频哪醇酯发生偶联反应生成中间体40, 中间体40与不同取代的溴苯化合物发生Suzuki偶联反应得到目标化合物28和29。具体操作步骤见后文实验部分。
| Table 1 Physical constants and ESI of compounds 6-29 |
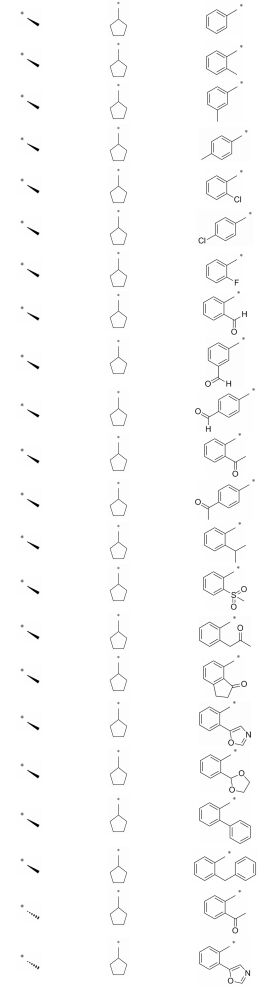
| Table 2 The NMR data of target compounds 6-29 |
目标化合物6 ~29对BRD4(Ⅰ)抑制活性见表 3。采用FA法对24个化合物进行了初步活性测定, 结果表明大部分化合物对BRD4(Ⅰ)都有抑制活性, 尤其是化合物16、22、28和29, 它们对BRD4(Ⅰ)的IC50小于100 nmol·L-1。
| Table 3 The inhibitory activities of compounds 6 -29 on BRD4-BD1 by fluorescence anisotropy assay. aAverage value by two independent experimental measurements; bmean ± SD (n = 2); cN.D.: Not detection |
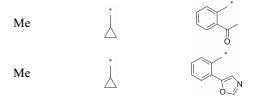
通过比较化合物6 ~12发现在侧链苯环邻位引入甲基、氟、氯或对位引入甲基、氯或间位引入甲基都不能明显改善化合物的分子活性, 它们在1 μmol·L-1时对BRD4(Ⅰ)的抑制率都很低。当在侧链苯环邻、间或对位引入甲酰基时化合物对BRD4(Ⅰ)抑制活性明显提高(比较化合物13 ~15与6), 当在侧链苯环邻位用乙酰基替换甲酰基时化合物的酶活性提高约6倍(比较化合物16与13), 而当在侧链苯环对位用乙酰基替换甲酰基时化合物的酶活性提高约1.9倍(比较化合物17与15)。
根据初步构效关系可知, 在侧链苯环邻位引入乙酰基时化合物的活性显著提高, 所以接下来在苯环邻位进行进一步探索, 希望能找到活性更好的化合物(表 3)。比较化合物18与16可以发现, 当去除苯环邻位乙酰基的羰基氧时化合物的分子活性显著降低, 说明这个位置的羰基氧可能形成关键氢键作用, 对活性影响很大。比较化合物19 ~21与16可以发现, 当用甲砜基或丙酮基替换乙酰基或者把乙酰基与侧链苯环环化时化合物的分子活性都明显降低, 尤其是化合物19和20, 对BRD4(Ⅰ)的IC50降低至约1 μmol·L-1。比较化合物22与16可以发现, 在侧链苯环邻位用噁唑基替换乙酰基时化合物的活性稍微提高一些, 而当用1, 3-二氧戊环、苯基或者苄基(23 ~25)替换乙酰基时化合物的分子活性相对于化合物16显著降低, 尤其是化合物25, 其分子活性完全丧失。接下来保留侧链苯环邻位是噁唑基或乙酰基而改变母核结构, 希望能找到活性更好的化合物。首先只改变母核C3位置的手性, 将其由R型换成S构型, 如化合物26和27, 其对BRD4(Ⅰ)的活性大大降低, 说明母核C3位置手性对活性影响很大。接下来把二氢喹喔啉-2(1H)-酮母核上的环戊基替换成环丙基以及用消旋体替换C3位置的R构型合成化合物28和29, 发现它们对BRD4(Ⅰ)的抑制活性可以保持(比较化合物28和16或比较化合物29和22)。

|
Scheme1 Synthetic routes of compounds 6 -25. Reagents and conditions: a) D-Alanine, K2CO3, EtOH, H2O, 80 ℃, 8 h; b) K2CO3, Na2S2O4, H2O, 60 ℃, overnight; c) Phenylsilane, cyclopentanone, dibutyltin dichloride, THF, rt, 10 h; d) NaH, DMF, 0 ℃, 30 min, iodomethane, rt, 2 h; e) Pd(dppf)Cl2, bis(pinacolato)diboron, AcOK, dioxane, 80 ℃, overnight; f) Pd(dppf)Cl2, NaHCO3, THF, H2O, 80 ℃, overnight |

|
Scheme2 Synthetic routes of compounds 28 -29. Reagents and conditions: a) Cyclopropylamine, ClCH2CH2Cl, 80 ℃, 12 h; b) Fe, NH4Cl, EtOH, 80 ℃, 1 h; c) 1. 2-Bromopropanoyl bromide, DIPEA, DCM, 0 ℃-rt, 2 h; 2. CH3CN, DIPEA, 80 ℃, overnight; d) NaH, DMF, 0 ℃, 30 min, iodomethane, rt, 2 h; e) Pd(dppf)Cl2, bis(pinacolato)diboron, AcOK, Dioxane, 80 ℃, overnight; f) Pd(dppf)Cl2, NaHCO3, THF, H2O, 80 ℃, overnight |
根据BI-2536与BRD4和PLK1不同的结合模式, 用二氢喹喔啉-2(1H)-酮代替BI-2536中的7, 8-二氢蝶啶-6-(5H)-酮来设计新颖结构的BRD4抑制剂。通过叠合比较化合物3与BRD4-BD1的共晶结构和本课题组之前报道的一类2-噻唑酮类化合物5与BRD4-BD1的共晶结构, 推测在二氢喹喔啉-2(1H)-酮母核的C6位置增加取代基可能引入基团与WPF亚口袋形成范德华作用。通过探索新的二氢喹喔啉-2(1H)-酮骨架的构效关系, 最终获得一类结构新颖的具有苯基侧链的BRD4抑制剂。其中在侧链苯环邻位引入乙酰基和噁唑基时化合物的分子活性最好, 如化合物16、22 、28和29, 它们对BRD4(Ⅰ)的抑制活性比临床二期的I-BET762活性略优。
实验部分实验中所用的试剂及溶剂均为商品试剂, 除有特别注明外, 所有试剂和溶剂均直接使用, 未作进一步处理。柱色谱硅胶(200~300目)为青岛海洋化工厂生产; HSGF-254型薄层色谱板及厚合成板为烟台江友硅胶开发有限公司生产, 薄层色谱板采用紫外灯检测或碘缸显色。1H NMR, 13C NMR: Bruker AVANCE Ⅲ-400型、AVANCE Ⅲ-500型以及Varian Mercury-300型核磁共振仪, TMS为内标; ESI-MS: Finnigan LCQ-Deca XP型液相色谱-质谱联用仪; HPLC: Gilson Nebula Series高效液相色谱仪; 分析柱: YMC CombiScreen ODS-A (50 mm × 4.6 mm, 5 µm)。
1 化合物的合成 1.1 (5-溴-2-硝基苯基)-D-丙氨酸(31)将4-溴-2氟硝基苯(25 g, 113.64 mmol)、D-氨基丙酸(11.12 g, 125.0 mmol)和碳酸钾(17.25 g, 125.0 mmol)溶于500 mL乙醇-水=3:1的混合溶剂中, 加热回流8 h, 用TLC板监测, 反应完后冷却至室温, 用1 mol·L-1盐酸调节pH至2~3, 将固体过滤, 用200 mL石油醚洗涤固体, 干燥得28.7 g化合物31, 为黄色固体, 收率88%。1H NMR (400 MHz, CDCl3) δ 8.35 (d, J = 6.9 Hz, 1H), 8.06 (d, J = 9.1 Hz, 1H), 6.90 (s, 1H), 6.85 (d, J = 9.2 Hz, 1H), 4.33 (p, J = 7.0 Hz, 1H), 1.67 (d, J = 7.0 Hz, 3H)。
1.2 (R)-6-溴-3-甲基-3, 4-二氢喹喔啉-2(1H)-酮(32)将化合物31 (28.7 g, 99.31 mmol)和K2CO3 (27.41 g, 198.62 mmol)溶于500 mL水中, 分批加入连二亚硫酸钠(86.45 g, 496.55 mmol), 60 ℃反应过夜。产生大量沉淀, 用TLC板监测, 反应完成后将固体过滤, 用300 mL水洗涤, 干燥得9 g化合物32, 为黄色固体, 收率38%。1H NMR (400 MHz, CDCl3) δ 9.42 (s, 1H), 6.89~6.83 (m, 1H), 6.81 (s, 1H), 6.68~6.59 (m, 1H), 4.02 (q, J = 6.7 Hz, 1H), 3.92 (s, 1H), 1.45 (d, J = 6.7 Hz, 3H)。
1.3 (R)-6-溴-4-环戊基-3-甲基-3, 4-二氢喹喔啉-2(1H)-酮(33)将化合物32 (9 g, 37.34 mmol)、苯硅烷(11.90 g, 113.14 mmol)、环戊酮(10.0 mL, 113.14 mmol)以及二丁基二氯化锡(17.02 g, 56.01 mmol)溶于100 mL四氢呋喃中, 常温搅拌10 h, 用TLC板监测, 反应完后将溶剂蒸干, 有机相硅胶拌样通过色谱柱纯化, 使用乙酸乙酯/石油醚= 0~30%梯度洗脱, 得10.4 g化合物33, 为棕色油状, 收率90%。1H NMR (400 MHz, CDCl3) δ 9.68 (s, 1H), 6.92 (d, J = 1.9 Hz, 1H), 6.88 (dd, J = 8.2, 2.0 Hz, 1H), 6.69 (d, J = 8.2 Hz, 1H), 4.10 (q, J = 6.8 Hz, 1H), 3.88~3.75 (m, 1H), 2.08~1.94 (m, 2H), 1.78~1.55 (m, 6H), 1.14 (d, J = 6.8 Hz, 3H); MS (ESI) [M+H]+: 309.03。
1.4 (R)-6-溴-4-环戊基-1, 3-二甲基-3, 4-二氢喹喔啉-2(1H)-酮(34)将NaH (1.61 g, 67.27 mmol)悬浮于100 mL干燥的DMF中, 在冰浴下搅拌5 min, 加入80 mL化合物33 (10.4 g, 33.63 mmol)的DMF溶液, 搅拌20 min, 缓慢滴入碘甲烷(3.14 mL, 50.45 mmol), 将体系移至室温反应4 h, 用TLC板监测, 反应完后在冰浴下加水淬灭, 加入300 mL水, 用乙酸乙酯萃取两次, 合并有机相, 用600 mL饱和食盐水洗一次, 无水硫酸钠干燥, 蒸干溶剂, 有机相硅胶拌样通过色谱柱纯化, 使用乙酸乙酯/石油醚=0~25%梯度洗脱, 得10 g化合物34, 为浅黄色液体, 收率92%。1H NMR (400 MHz, CDCl3) δ 6.97 (dd, J = 8.4, 2.1 Hz, 1H), 6.92 (d, J = 2.1 Hz, 1H), 6.78 (d, J = 8.5 Hz, 1H), 4.17 (q, J = 6.8 Hz, 1H), 3.81~3.72 (m, 1H), 3.33 (s, 3H), 2.08~1.96 (m, 2H), 1.84~1.74 (m, 1H), 1.72~1.58 (m, 5H), 1.05 (d, J = 6.8 Hz, 3H)。
1.5 (R)-4-环戊基-1, 3-二甲基-6-(4, 4, 5, 5-四甲基-1, 3, 2-二氧杂硼杂环戊烷-2-基)-3, 4-二氢喹喔啉-2(1H)-酮(35)将化合物34 (10 g, 30.96 mmol)、联硼酸频哪醇酯(8.65 g, 34.06 mmol)以及醋酸钾(6.08 g, 61.92 mmol)溶于400 mL DMSO中, 通氩气10 min, 加入[1, 1'-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物(1.26 g, 1.55 mmol), 继续通气2 min, 氩气保护下加热至80 ℃回流过夜, 用TLC板监测, 反应完后用二氯甲烷(400 mL × 2) 和400 mL水萃取, 合并有机层, 用800 mL饱和食盐水洗涤, 无水硫酸钠干燥, 有机相硅胶拌样通过色谱柱纯化, 使用乙酸乙酯/石油醚= 0~10%梯度洗脱, 得9.5 g化合物35, 为浅黄色固体, 收率83%。1H NMR (400 MHz, CDCl3) δ 7.36 (d, J = 8.3 Hz, 1H), 7.29 (s, 1H), 6.95 (d, J = 7.9 Hz, 1H), 4.17 (q, J = 6.8 Hz, 1H), 3.94~3.85 (m, 1H), 3.37 (s, 3H), 2.09~1.98 (m, 2H), 1.83~1.74 (m, 1H), 1.73~1.58 (m, 5H), 1.35 (s, 12H), 1.03 (d, J = 6.9 Hz, 3H); MS (ESI) [M+H]+: 371.14。
1.6 目标化合物6的合成将化合物35(0.1 g, 0.27 mmol)、溴苯(0.056 mL, 0.54 mmol)以及饱和碳酸氢钠(45 mg, 5.40 mmol)溶液溶于2 mL THF中, 通氩气10 min, 加入[1, 1'-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物(22 mg, 0.027 mmol), 继续通气2 min, 氩气保护下加热至80 ℃回流过夜, 用TLC板监测, 反应完后用二氯甲烷(40 mL × 2) 和40 mL水萃取, 用80 mL饱和食盐水洗涤, 无水硫酸钠干燥, 有机相硅胶拌样通过色谱柱纯化, 使用乙酸乙酯/石油醚=0~25%梯度洗脱, 得0.06 g化合物6, 为浅黄色液体, 收率69%。
1.7 5-溴-N-环丙基-2-硝基苯胺(36)4-溴-2-氟-1-硝基苯(10 g, 45.45 mmol)溶于30 mL1, 2-二氯乙烷中, 加入环丙胺(6.29 mL, 90.91 mmol), 然后80 ℃回流12 h, 用TLC监测反应, 反应完后蒸干溶剂, 用二氯甲烷(40 mL × 2) 和40 mL水萃取, 合并有机层, 用80 mL饱和食盐水洗涤, 有机相无水硫酸钠干燥, 有机相蒸干得10.2 g化合物36, 为红色固体, 产率87%。1H NMR (400 MHz, CDCl3) δ 8.05 (s, 1H), 7.96 (d, J = 9.1 Hz, 1H), 7.45 (d, J = 2.0 Hz, 1H), 6.77 (dd, J = 9.1, 2.0 Hz, 1H), 2.60~2.50 (m, 1H), 0.97~0.88 (m, 2H), 0.69~0.61 (m, 2H)。
1.8 5-溴-N-环丙基苯-1, 2-二胺(37)化合物36 (10 g, 38.90 mmol)溶于25 mL乙醇中, 加入氯化铵溶液(194.55 mmol, 10 mL H2O), 再加入铁粉(10.89 g, 194.55 mmol), 80 ℃反应1 h, 用TLC监测反应, 反应完之后用硅藻土滤去铁粉, 用乙酸乙酯(40 mL × 2) 和40 mL水萃取, 合并有机层, 用80 mL饱和食盐水洗涤, 有机相无水硫酸钠干燥, 有机相蒸干得5.2 g化合物37, 为棕色液体, 产率59%。1H NMR (400 MHz, CDCl3) δ 7.13 (d, J = 2.1 Hz, 1H), 6.79 (dd, J = 8.1, 2.1 Hz, 1H), 6.53 (d, J = 8.1 Hz, 1H), 3.59~3.03 (m, 2H), 2.45~2.32 (m, 1H), 0.76 (q, J = 6.4 Hz, 2H), 0.57~0.44 (m, 2H)。
1.9 6-溴-4-环丙基-3-甲基-3, 4-二氢喹喔啉-2(1H)-酮(38)化合物37 (5.2 g, 22.90 mmol)溶于干燥的15 mL二氯甲烷中, 冷却至0 ℃, 加入N, N-二异丙基乙胺(7.98 mL, 45.80 mmol)和2-溴丙酰溴(3.12 mL, 29.77 mmol), 然后室温反应2 h, 用TLC监测反应, 反应完后用二氯甲烷(40 mL × 2) 和40 mL水萃取, 合并有机层, 用80 mL饱和食盐水洗涤, 有机相无水硫酸钠干燥, 有机相蒸干得中间体, 该中间体溶于20 mL乙腈中, 加入8 mL DIPEA, 然后80 ℃反应过夜, 用TLC监测反应, 反应完后蒸干溶剂, 用二氯甲烷(40 mL × 2) 和40 mL水萃取, 合并有机层, 用80 mL饱和食盐水洗涤, 有机相无水硫酸钠干燥, 蒸干并用硅胶拌样通过色谱柱纯化, 使用乙酸乙酯/石油醚= 0~30%梯度洗脱, 得到2 g化合物38, 为浅黄色固体, 产率31%。1H NMR (400 MHz, CDCl3) δ 9.10 (s, 1H), 7.18 (d, J = 1.8 Hz, 1H), 6.91 (dd, J = 8.2, 1.9 Hz, 1H), 6.64 (d, J = 8.2 Hz, 1H), 4.04 (q, J = 6.8 Hz, 1H), 2.46~2.36 (m, 1H), 1.24 (d, J = 6.9 Hz, 3H), 1.04~0.96 (m, 1H), 0.85~0.77 (m, 1H), 0.68~0.60 (m, 1H), 0.60~0.52 (m, 1H); LCMS m/z (ESI, positive) found [M+H]+ 281.17。
1.10 6-溴-4-环丙基-1, 3-二甲基-3, 4-二氢喹喔啉-2(1H)-酮(39)化合物38 (2 g, 7.11 mmol)溶于8 mL DMF中, 冷却至0 ℃,加入氢化钠(0.52 g, 21.34 mmol), 0 ℃反应0.5 h后加入碘甲烷(0.67 mL, 10.67 mmol), 然后室温反应1 h, 用TLC监测反应, 反应完后冷却至0 ℃, 加入稀盐酸调pH至7~8, 然后用二氯甲烷(20 mL × 2) 和20 mL水萃取, 合并有机层, 用40 mL饱和食盐水洗涤, 有机相无水硫酸钠干燥, 蒸干并用硅胶拌样通过色谱柱纯化, 使用乙酸乙酯/石油醚= 0~25%梯度洗脱, 得到1.8 g化合物39, 产率86%。1H NMR (400 MHz, CDCl3) δ 7.14 (d, J = 2.2 Hz, 1H), 6.91 (dd, J = 8.5, 2.1 Hz, 1H), 6.70 (d, J = 8.5 Hz, 1H), 4.03 (q, J = 6.9 Hz, 1H), 3.24 (s, 3H), 2.37~2.28 (m, 1H), 1.10 (d, J = 6.9 Hz, 3H), 0.97~0.90 (m, 1H), 0.79~0.68 (m, 1H), 0.62~0.52 (m, 1H), 0.50~0.42 (m, 1H); LCMS m/z (ESI, positive) found [M+H]+ 295.15。
1.11 4-环丙基-1, 3-二甲基-6-(4, 4, 5, 5-四甲基-1, 3, 2-二氧杂环戊硼烷-2-基)-3, 4-二氢喹喔啉-2(1H)-酮(40)将化合物39 (1.8 g, 6.10 mmol)、联硼酸频哪醇酯(2.32 g, 9.15 mmol)以及醋酸钾(1.20 g, 12.2 mmol)溶于5 mL DMSO中, 通氩气10 min, 加入[1, 1'-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物(0.50 g, 0.61 mmol), 继续通气2 min, 氩气保护下加热至80 ℃回流过夜, 用TLC板监测, 反应完后用二氯甲烷(40 mL × 2) 和40 mL水萃取, 合并有机层, 用80 mL饱和食盐水洗涤, 无水硫酸钠干燥, 有机相硅胶拌样通过色谱柱纯化, 使用乙酸乙酯/石油醚= 0~10%梯度洗脱, 得1.5 g化合物40, 为浅黄色固体, 收率72%。
1.12 目标化合物28的合成将化合物40 (0.1 g, 0.29 mmol)、1-(3-溴苯基)乙-1-酮(0.12 g, 0.58 mmol)以及饱和碳酸氢钠(49 mg, 0.58 mmol)溶液溶于2 mL THF中, 通氩气10 min, 加入[1, 1'-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物(24 mg, 0.029 mmol), 继续通气2 min, 氩气保护下加热至80 ℃回流过夜, 用TLC板监测, 反应完后用二氯甲烷(20 mL × 2) 和20 mL水萃取, 用40 mL饱和食盐水洗涤, 无水硫酸钠干燥, 有机相硅胶拌样通过色谱柱纯化, 使用乙酸乙酯/石油醚= 0~25%梯度洗脱, 得0.07 g化合物28, 为浅黄色液体, 收率72%。
2 BRD4(Ⅰ)识别结构域的表达和纯化方法通过检索文献[5]和PDB数据库系统, 确定需表达的序列, 采用His6标签, 以利于蛋白纯化BRD4蛋白BRD4(Ⅰ)结构域。表达的载体为PET28a, 采用大肠杆菌表达系统。
3 溴结构域识别蛋白BRD4抑制剂酶活性测试方法采用荧光各异向性测试方法(fluorescence anisotropy, FA)测试化合物与BRD4(Ⅰ)的结合活性。具体测试原理和操作步骤参见我们课题组赵乐乐之前发表的文献[18]。
| [1] | Younus I, Reddy DS. Epigenetic interventions for epilepto-genesis:a new frontier for curing epilepsy[J]. Pharmacol Ther, 2017, 177: 108–122. DOI:10.1016/j.pharmthera.2017.03.002 |
| [2] | Hojfeldt JW, Agger K, Helin K. Histone lysine demethy-lases as targets for anticancer therapy[J]. Nat Rev Drug Discov, 2013, 12: 917–930. DOI:10.1038/nrd4154 |
| [3] | Tamkun JW, Deuring R, Scott MP, et al. brahma:a regula-tor of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2/SWI2[J]. Cell, 1992, 68: 561–572. DOI:10.1016/0092-8674(92)90191-E |
| [4] | Filippakopoulos P, Picaud S, Mangos M, et al. Histone recognition and large-scale structural analysis of the human bromodomain family[J]. Cell, 2012, 149: 214–231. DOI:10.1016/j.cell.2012.02.013 |
| [5] | Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibi-tion of BET bromodomains[J]. Nature, 2010, 468: 1067–1073. DOI:10.1038/nature09504 |
| [6] | Sahai V, Redig AJ, Collier KA, et al. Targeting BET bromo-domain proteins in solid tumors[J]. Oncotarget, 2016, 7: 53997–54009. DOI:10.18632/oncotarget.v7i33 |
| [7] | Dhalluin C, Carlson JE, Zeng L, et al. Structure and ligand of a histone acetyltransferase bromodomain[J]. Nature, 1999, 399: 491–496. DOI:10.1038/20974 |
| [8] | Sanchez R, Zhou MM. The role of human bromodomains in chromatin biology and gene transcription[J]. Curr Opin Drug Discov Dev, 2009, 12: 659–665. |
| [9] | Filippakopoulos P, Knapp S. Targeting bromodomains:epigenetic readers of lysine acetylation[J]. Nat Rev Drug Discov, 2014, 13: 337–356. DOI:10.1038/nrd4286 |
| [10] | Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition[J]. Mol Cell, 2014, 54: 728–736. DOI:10.1016/j.molcel.2014.05.016 |
| [11] | Kuzume T, Kubonishi I, Takeuchi S, et al. Establishment and characterization of a thymic carcinoma cellline (Ty-82) carrying t(15;19)(q15;p13) chromosome abnormality[J]. Int J Cancer, 1992, 50: 259–264. DOI:10.1002/(ISSN)1097-0215 |
| [12] | French CA, Miyoshi I, Kubonishi I, et al. BRD4-NUT fusion oncogene:a novel mechanism in aggressive carcinoma[J]. Cancer Res, 2003, 63: 304–307. |
| [13] | Nicodeme E, Jeffrey KL, Schaefer U, et al. Suppression of inflammation by a synthetic histone mimic[J]. Nature, 2010, 468: 1119–1123. DOI:10.1038/nature09589 |
| [14] | Martin MP, Olesen SH, Georg GI, et al. Cyclin-dependent kinase inhibitor dinaciclib interacts with the acetyl-lysine recognition site of bromodomains[J]. ACS Chem Biol, 2013, 8: 2360–2365. DOI:10.1021/cb4003283 |
| [15] | Knapp S, Arruda P, Blagg J, et al. A public-private partner-ship to unlock the untargeted kinome[J]. Nat Chem Biol, 2013, 9: 3–6. |
| [16] | Ember SW, Zhu JY, Olesen SH, et al. Acetyl-lysine binding site of bromodomain-containing protein 4(BRD4) interacts with diverse kinase inhibitors[J]. ACS Chem Biol, 2014, 9: 1160–1171. DOI:10.1021/cb500072z |
| [17] | Chen LJ, Yap JL, Yoshioka M, et al. BRD4 struc-ture-activity relationships of dual PLK1 kinase/BRD4 bromodomain inhibitor BI-2536[J]. ACS Med Chem Lett, 2015, 6: 764–769. DOI:10.1021/acsmedchemlett.5b00084 |
| [18] | Zhao LL, Cao DY, Chen TT, et al. Fragment-based drug discovery of 2-thiazolidinones as inhibitors of the histone reader BRD4 bromodomain[J]. J Med Chem, 2013, 56: 3833–3851. DOI:10.1021/jm301793a |