 2017, Vol. 52
2017, Vol. 52

编者按
新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
分子和细胞生物学研究揭示了刺猬信号通路在生命过程的作用和某些癌症的发生机制。但由认识异常的蛋白与受体的作用过渡到新药的创制, 小分子抑制剂与受体靶标的成药性, 贯穿于研发的全过程。构建维莫德吉的化学结构, 是在修改骨架、更换片段、变换基团的分子操作中, 提高了生物活性, 完善了成药性, 最后成为抗基底细胞癌的首创性药物, 同时也确证了刺猬信号通路是药物作用的靶标。维莫德吉在药物研究中的地位可用FDA的专家帕兹杜尔的评价加以诠释, 他说: “我们对涉及肿瘤的信号通路(如刺猬通路)的理解, 使研发治疗特殊疾病的靶向药物成为可能, 这种方法变得越来越普通, 并有可能使抗肿瘤药物研发更快。这对可获得更有效、不良反应少的治疗的患者很重要”。
在动物发育的过程中, 刺猬蛋白(hedgehog, HH)信号通路是重要的调控因子, 某些癌症的发生也与hedgehog通路异常有关。刺猬通路名称的起源, 是果蝇的刺猬蛋白基因若发生突变, 其胚胎呈多毛团状, 酷似受惊的刺猬, 故而得名。
哺乳动物中存在3个hedgehog同源基因: Sonic hedgehog (Shh)、Indian hedgehog (Ihh)和Desert hedgehog (Dhh), 分别编码SHH、IHH和DHH蛋白。HH蛋白由两个结构域组成:氨基端结构域(HH-N)及羧基端结构域(HH-C), 其中HH-N有HH蛋白的信号活性, 而HH-C具有自身蛋白水解酶活性及胆固醇转移酶功能。HH前体蛋白在内质网中通过自身催化分裂成HH-N及HH-C两部分, 其中HH-C共价结合胆固醇分子、并将其转移到HH-N的羧基端, 随后在酰基转移酶的作用下HH-N氨基端的半胱氨酸发生棕榈酰化。HH蛋白只有通过这些翻译后的修饰过程才能获得完全的功能。HH信号传递受靶细胞膜上两种受体Patched (PTCH)和Smoothened (SMO)的控制。受体PTCH是由12个跨膜区的肽链构成, 是HH蛋白的受体, 却对HH信号起负调控作用。受体SMO由7个跨膜区的单一肽链构成。
在正常情况下, PTCH抑制SMO蛋白活性, 从而抑制下游的通路, 这时下游的Gli蛋白在蛋白酶体内被截断, 并以羧基端被截断的形式进入细胞核内, 抑制下游靶基因的转录。当PTCH和HH结合以后, 解除对SMO的抑制作用, 使得全长Gli蛋白进入核内, 激活下游靶基因转录。若PTCH发生突变或缺失, 或者SMO发生突变, 则对PTCH的抑制作用不敏感, 使基因活化, 导致HH信号通路失控, 使Gli持续激活, 启动靶基因转录。图 1是hedgehog信号通路的活化和抑制剂作用的示意图。

|
图 1 Hedgehog信号通路的活化和抑制剂作用的示意图 |
一些癌症如基底细胞癌的发生和发展与异常的hedgehog信号通路密切相关。以SMO受体为靶标是研制这类抗肿瘤药的重要环节。
2 活性评价方法评价化合物对SMO受体的抑制活性, 是用小鼠胚胎成纤维细胞。成纤细胞中装有Gli结合位点的荧光素酶报告基因下游的质粒, 用SHH刺激细胞, 经光学方法测定荧光素酶的活性。当SMO受体被拮抗剂阻断, 通过测定该光学信号的下降值, 计算化合物抑制细胞的IC50。
3 苗头化合物最初发现从北美山藜芦(Veratrum californicum)分离的天然产物环帕胺(1, cyclopamine)具有抑制SMO受体、可阻断Hedgehog通路, 但环帕胺有强致畸作用, 且结构复杂, 不能作为先导物。

|
Genetech公司用随机筛选方法, 发现曾为研发趋化因子受体而合成的喹唑啉酮化合物2对SMO受体有弱抑制作用, IC50 = 1.4 μmol·L-1。2作为苗头化合物分子较大, 相对分子质量为586, 含非氢原子35个, 配体效率LE = 0.23, 反映了分子中存在无用的冗余原子, 缺少必需的药效团因素。为了将2改造成先导物, 合成了保留两个环的化合物3和4, 经评价发现活性显著下降, 在10 μmol·L-1浓度下未呈现抑制作用, 提示被切割掉的苯胺甲酰片段(即含有脲连接基)是不容去除的, 说明总体结构框架不能简约。
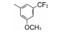
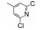
3.2 结构的微调苗头化合物的骨架既然不能大动, 但由于化合物的合成容易实现, 因而可以通过合成不同结构的化合物, 探索构效关系。首先保持分子右侧的R3不变, 设计合成了去除甲基的两个化合物5和6。发现去除两个甲基的化合物5比2的活性提高1倍, 但去除一个甲基的6活性反而降低, 活性大约是2的30%, 提示R1和R2宜为氢原子。进而对R3加以变换, 合成了有代表性的化合物7~9, 其中9的活性最强。表 1列出了这些化合物的结构和抑制活性。
| 表 1 化合物2和5~9的结构和抑制SMO受体的活性 |



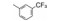

Genetech公司还发现骨架相近的含有氯代苯并噻吩的化合物对SMO受体具有(相反的)激动作用, 经结构改造获得了活性较高的激动剂10, EC50 = 3 nmol·L-1 (Baxter AD, Boyd EA, Guicherit OM, et al. Small organic molecule regulators of cell proliferation. PCT Int. Appl., 2001, WO 2001-US10296)。激动剂10作为工具药对于揭示信号通路和作用机制有重要意义。进而发现了含有苯并咪唑的化合物11具有阻止激动剂结合SMO受体的活性, IC50 = 30 nmol·L-1 (Chen JK, Taipale J, Young KE, et al. Small molecule modulation of smoothened activity. Proc Nat Acad Sci USA, 2002, 99: 14071–14076)。
4 先导化合物的优化 4.1 拼合优势结构因素化合物11具有较强的抑制SMO受体的活性, 化学结构骨架也比较简约; 前述的化合物8的吡啶环和9的苯环上取代基对提高活性有利, 遂将这些有利的结构因素组合在一起, 设计合成了化合物12, 12具有较强的活性(IC50 = 12 nmol·L-1), 是个里程碑式的化合物。

|

|
然而化合物12在成药性上有两个缺点, 一是较低的代谢稳定性, 用犬和人肝微粒体温孵, 可被氧化代谢, 预测的清除率较高, 分别为22和9.4 mL·min-1·kg-1; 另一缺点是溶解性差, 在pH 6.4的水溶解度仅为0.3 μg·mL-1, 所以不能成为候选化合物。在结构改造中合成联(苯并咪唑)-氯苯的难度也较大。右边的酰胺片段的合成相对容易, 因而首先对苯并咪唑环加以变换, 以尽快优化出活性骨架。表 2列出了用各种芳环替换苯并咪唑的结构和活性。
| 表 2 化合物12、13~31的结构和活性 |
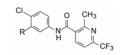




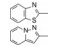
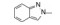
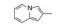




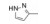

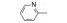


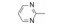

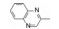
分析芳环的变换与活性的关系, 内容如下: ① 与氯苯连键的邻位(即迫位)须有氢键接受体的原子或基团, 如化合物12的N3原子, 12的电子等排体(13)由于吲哚环缺少氢键接受体因素(吲哚的N-H只是氢键给体), 活性下降了40倍, 12的N-甲基化合物14, 两个氮原子都是氢键接受体, 活性与12相同。苯并噻唑16和喹喔啉31也因此有较强的活性。② 邻位氮原子若碱性弱, 难于形成氢键, 导致活性下降, 例如吡唑并吡啶17和吲唑18的碱性弱于咪唑化合物, 活性因此减弱。单环吡唑23有中等强度活性, 而其异构体24活性很低, 这是因为24存在互变异构现象, 降低了接受氢键的能力。但24经N-甲基化得到的25, 因阻止了互变异构, 活性与23相近。③ 单环2-吡啶化合物26的活性很高IC50 = 42 nmol·L-1, 将氮原子移至3或4位, 27和28的活性很低, 也是因为缺少邻位氮原子的缘故。2-吡啶基结构小而简单, 是个优选的结构片段。④ 非杂环的苯胺甲酰21也有中等强度活性, 推测羰基氧为氢键接受体。
在这些优化的分子中, 14具有最高的活性(IC50 = 9 nmol·L-1), 不过它对CYP2C9有中等强度的抑制作用(IC50 = 1.5 μmol·L-1), 而且代谢清除率较快, 比化合物12和26快4~6倍。所以, 成药性的缺陷使化合物14不能成为候选物。化合物31因溶解性低影响了吸收和成药性。
4.3 芳胺的变换在以苯并咪唑为母核的系列化合物中, 结构右侧的芳酰基是取代的烟酸时活性最强, 例如化合物26。下一步探索的是若用结构简约的吡啶替换苯并咪唑(如化合物14), 右侧的构效关系可能发生新的变化。为此合成了以联吡啶-氯苯为母核的化合物32~42, 考察右侧酰基变换的构效关系, 表 3中列出化合物的结构和活性。
| 表 3 化合物26、32~42的结构和活性 |
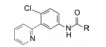


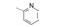
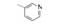

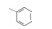

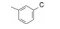
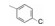
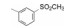

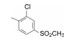
构效关系的要点总结如下: ① 右侧吡啶环上的氮原子并非必需。将化合物26吡啶环上的2'-甲基和4'-三氟甲基去除, 无取代的2-吡啶甲酸(33)、烟酸(34)和异烟酸(35)的活性虽然下降, 但与苯甲酸化合物36的活性相近, 提示该氮原子没有参与受体的结合作用。② 芳环2-位取代基的贡献是显著的。2-甲基或2-氯取代可提高活性, 推测可能有两个因素起作用:甲基或氯原子的亲脂性, 有助于化合物与受体的疏水腔发生疏水-疏水相互作用; 也可能是2-位基团的阻迫效应使芳环与酰胺平面的夹角增大, 致使稳固的芳环-酰胺近于正交的构象有利于同受体的结合(根据对剑桥数据库的统计, 苯甲酰胺的苯基与酰胺的平面夹角为30 /150 , 2-氯苯甲酰胺的夹角为60 /120 )。2-氯苯甲酰胺化合物37的IC50 = 110 nmol·L-1, 比无取代的苯甲酰胺活性提高7倍。若将氯原子移到3-或4-位, 解除了阻迫效应, 活性显著下降, 提示氯在2位的重要性。③ 芳环上引入吸电子基团如甲磺酰基, 可提高活性, 而且4-位优于3-位取代, 如化合物41比40的活性高2倍。④ 将2-氯和4-甲磺酰基的优势取代集合于苯甲酸上, 合成的化合物42具有很高的活性(IC50 = 13 nmol·L-1)。
5 高活性化合物的综合比较—候选物的确定由联(苯并咪唑)-苯胺-烟酰胺为骨架的起始化合物, 优化成联2-吡啶-苯胺-2-氯苯甲酰胺类化合物, 主要是以对细胞内hedgehog通路的SMO受体的抑制活性作为评价标准的, 其间虽然也考虑到化合物的代谢稳定性、对CYP的抑制作用, 以及溶解性能等性质, 但并没有对活性较高的化合物在成药性上作系统的比较。下一步是对高活性化合物作药代和物化性质的比较, 表 4列出了6个高活性化合物的一些代谢参数和物化性质, 可以看出化合物42具有明显的优势:很高的抑制SMO受体的活性, 在哺乳动物体内有较低的清除率, 以及水溶解性较好等(Robarge KD, Brunton SA, Castanedo GM, et al. GDC-0449-a potent inhibitor of the hedgehog pathway. Bioorg Med Chem Lett, 2009, 19: 5576-5581)。

|
| 表 4 有代表性化合物的活性、药代和物化性质的比较 |


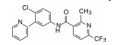
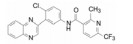
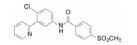

进一步还用移植依赖于hedgehog通路的髓母细胞瘤的裸鼠评价了化合物26、41和42的抑瘤作用, 也表明化合物42有显著作用。化合物41与42的区别是后者的端基有2-氯取代, 二者活性和药代性质相近, 41的分配系数logP值更适宜, 但溶解度显著低于42, 2-氯取代提高溶解性的原因可能是邻位效应阻止了苯环与酰胺共面性, 降低了分子间的晶格能。Genetech公司确定化合物42作为候选物, 定名为维莫德吉(vismodegib)进入临床研究, 于2012年经FDA批准成为首创的抗基底细胞癌药物, 用于不能手术或放疗的局部晚期皮肤基底细胞癌患者和肿瘤已转移的患者。