 2017, Vol. 52
2017, Vol. 52

2. 中国医学科学院、北京协和医学院药物研究所, 北京 100050

 , SHEN Zhu-fang2
, SHEN Zhu-fang2
2. Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100050, China
随着社会经济的发展、人们生活水平的提高和生活方式的改变, 糖尿病已经成为21世纪人类健康的最大威胁。2015年, IDF (International Diabetes Federation)统计数据显示4.15亿成年人患有糖尿病, 其中2型糖尿病占糖尿病患者95%以上, 且80%以上的2型糖尿病患者均伴有胰岛素抵抗。所以胰岛素增敏剂应该是治疗2型糖尿病的理想药物。噻唑烷二酮(thiazolidinedione, TZD)类药物是目前上市的唯一高度有效的治疗2型糖尿病的胰岛素增敏剂。1999年, GSK公司的罗格列酮(rosiglitazone, 商品名Avandia)、武田公司的吡格列酮(pioglitazone, 商品名Actos) (图 1)先后经美国食品药品监督管理局(US Food and Drug Administration, FDA)批准上市[1], 这类药物临床上不但能直击胰岛素抵抗, 而且显著地改善骨骼肌、脂肪组织及肝细胞的胰岛素敏感性、降低空腹高胰岛素及血糖水平, 预测临床应用前景良好[2-4]。但不幸的是, 相当数量的病人由于服用这类药出现充血性心衰、液体潴留、体重增加和骨质疏松等不良反应, 甚至有报道, 罗格列酮有增加心血管疾病的风险[5-6]。使这类唯一高度有效的胰岛素增敏剂的临床应用由于安全性问题而受到限制。

|
Figure 1 Rosiglitazone and pioglitazone |
过氧化酶体增殖物激活受体γ (peroxisome proliferator-activated receptor γ, PPARγ)是一种配体激活核转录因子, 在提高葡萄糖的耐受性和和胰岛素敏感性方面起着关键性作用[7-9], 被认为是TZDs药物的分子靶标。TZDs药物作为PPARγ完全激动剂, 以前的药物设计都是基于化合物对PPARγ的完全激动活性, 认为其激动活性越高, 对胰岛素增敏作用越强。但后来发现的PPARγ部分激动剂, 与PPARγ完全激动剂相比, 虽只部分激活转录基因, 却可以保持良好的降糖活性[10], 并降低完全激动剂导致的常见不良反应。如INT131 (图 2), 与完全激动剂罗格列酮相比, 虽然其激动活性较低, 但对胰岛素增敏活性和降糖效果相当, 骨质疏松等不良反应减小[11, 12]。因此人们对胰岛素增敏与PPARγ完全激动活性的相互关联提出了质疑。

|
Figure 2 Structure of INT131 and strategy of designing |
随着对降糖机制的不断探究, 后来的研究已表明, TZD类药物的胰岛素增敏作用是基于其对Cdk5介导的PPARγ Ser273的磷酸化的抑制, 起因于其对PPARγ的结合活性; 而TZD类药物的不良反应是起因于其对PPARγ的激动活性[13]。所以, 以PPARγ为靶标, 设计没有激动活性, 只有结合活性的新型PPARγ非激动配体, 研发只有增敏作用而无TZD类药物不良反应的治疗2型糖尿病新药成为目前的研究方向。
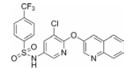
本文的研究正是基于报道的部分激动剂INT131的骨架结构, 以降低或去除其激动活性, 保持或提高其结合活性为目标, 参考INT131与PPARγ蛋白的复合物晶体结构信息[10], 对INT131结构的3个片段(A环、B环、C环)进行微调, 扰动INT131分子构象, 优化与PPARγ结合腔的匹配, 提高其结合活性与激动活性比值。
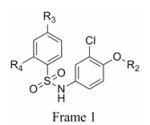
INT131分子中的2, 4-二氯苯基, 也是本文设计分子中R3和R4所在苯环(A环)位于螺旋7和螺旋3之间的通道内, 它的空间取向及与PPARγ蛋白的作用强弱对提高其结合活性与激动活性之比值可能有一定影响, 而且通道内A环有一定的延伸空间, 选择体积更大的三氟甲基来代替氯原子, 从而考察其对活性的影响。依据B片段的结构可分为苯环和吡啶环两个类型, 在B片段分别为苯环的单氯取代、双氯取代和吡啶环的单氯取代时, 通过对A片段和C片段的结构微调, 考察其对结合活性与激动活性之比值的影响。C片段是喹啉环结构, 最简单直接的调节是改为异喹啉和喹啉环中氮的氧化, 考察其对活性的影响。总之, 本文在保持INT131整个分子骨架结构不变的同时对A、B、C三部分进行结构优化(图 2), 试图寻找高结合能的PPARγ非激动配体, 为开发新型的治疗2型糖尿病的胰岛素增敏剂提供先导物结构。
结果与讨论 1 目标化合物的合成本文所设计的化合物合成路线也分为两部分, 一部分是以不同取代的硝基苯为原料, 另一部分以不同取代的硝基吡啶为原料经过取代、还原、缩合得到目标化合物(合成路线1)。经过实验探索发现:步骤a中, R2为3-羟基喹啉时采用THF为溶剂进行反应, 而R2为3-羟基异喹啉时则采用的溶剂是DMF, 若采用THF体系则不反应, 考虑可能是由于3-羟基异喹啉中氮原子与羟基形成的共轭体系, 导致氧的亲核性减弱, 只有在极性较大的溶剂体系内反应才可进行; 步骤b中, 硝基的还原需要严格控制反应条件, 在实验过程中, 尝试过锌粉和醋酸、氢气和钯碳进行还原, 但效果并不是太理想, 副产物较多。后来发现溶液加热到40 ℃再加入锌粉和氯化铵来还原硝基可以减少副反应的发生, 温度低的情况下加入还原剂, 硝基不能够一步转化为氨基, 可能生成一系列含有不同价态氮的化合物, 所以温度的控制是至关重要的; 步骤c中, 苯磺酰氯和胺的投料比控制在1.2:1以内, 否则未反应完的苯磺酰氯在柱色谱分离纯化过程中会影响产品的纯度, 进而影响收率, 另外锌粉与硝基苯的投料比应≧10, 否则反应不完全。化合物理化性质和核磁数据见表 1、2。

|
Scheme1 Synthsis of target compounds. Reagents and conditions: (a) R2-OH, Cs2CO3, THF or DMF, rt, 3 h; (b) CH3OH, H2O, Zn, NH4Cl, reflux, 30 min; (c) Benzene sulfonyl chloride, pyridine, DMAP, DCM, rt, overnight; (d) 3-Chloroperoxybenzoic acid, chloroform, rt, overnight |
| Table 1 Structures and physical property of the target compounds |
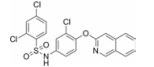
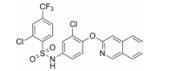

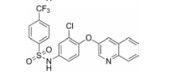

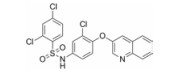
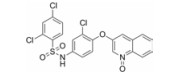
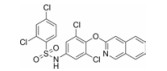
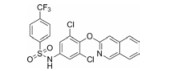




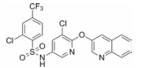
| Table 2 Spectral data of target compounds |
对合成的目标化合物分别进行细胞水平的PPARγ转录激动活性筛选和分子水平的PPARγ结合活性筛选。细胞水平的激动活性表示为:相对于阴性对照的受体激活倍数(Fold)和相对于阳性对照的活性百分比(%), 激动活性越高, 不良反应越大; 分子水平的结合活性表示:配体结合受体诱导下降的荧光强度, 相对于阴性对照的结合活性倍数(Fold)以及相对于阳性对照的活性百分比(%), 结合活性越高, 増敏活性越高; 从结合活性与激动活性的比值(A/T)考虑, 比值越高, 相对不良反应越低, 但要保持一定的结合活性或增敏活性才有意义。
三个结构骨架(骨架1、2、3) 及其活性筛选结果见表 3~5。
| Table 3 Activity of compounds 1-7 |
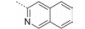
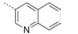
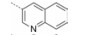
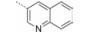

| Table 4 Activity of compounds 8-12 |
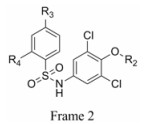

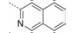
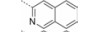


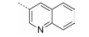
| Table 5 Activity of compounds 13-15 |
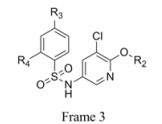


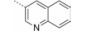
表 3结果表明, 与INT131的结构相比, 该部分化合物结构的B环由INT131的双氯取代苯环变为单氯取代苯环。R2为异喹啉环时, 与PPARγ的结合活性普遍较低, 但结合活性与激动活性比值均高于INT131, 其中R3=CF3、R4=H时结合活性及与激动活性之比值相对较高。R2为喹啉环时, 与PPARγ的结合活性普遍大幅升高, 但是结合活性及与激动活性之比值均比INT131略高, 其中R3=CF3、R4=Cl时结合活性及与激动活性之比值相对较高。R2为氮氧化的喹啉环时, 结合活性和激动活性均得到了提高, 但是激动活性提高的比例要高于结合活性。
表 4结果表明, 此骨架中B环结构与INT131相同。如果R2为喹啉环保持不变, R3=CF3、R4=H时, 结合活性和激动活性均有所降低, 但激动活性降低的比例要高于结合活性, 导致结合活性与激动活性比值相比INT131有了很大提升, 说明CF3更有利于激动活性的降低。R2为异喹啉环时, 结合活性和激动活性均大幅降低, 其比值也降低; R2为氮氧化的喹啉环时, 结合活性提高, 激动活性降低, 结合活性与激动活性比值提升了将近一倍。
表 5结果表明, 与INT131的结构相比, 该骨架结构的B环由INT131的双氯取代苯环变为单氯取代吡啶环。同样当R2为异喹啉环, R3=CF3、R4=H时结合活性和激动活性均大幅降低; 但R2为喹啉环时, 结合活性均较高, 与INT131相近; 而激动活性却降低很多, 尤其是化合物15, 激动活性与INT131相比, 降低10倍多, 为罗格列酮的1.41%, 其激动活性接近消失。
3 活性化合物的分子对接采用Accelrys公司Discovery Studio 2.5.5软件包中的Receptor-Ligand interactions模块下的Dock ligands (CDOCKER)程序进行了分子对接以及图形处理。INT131和化合物15分别与PPARγ (PDB: 3FUR)的对接结果见图 3。化合物15作为配体可以很好地进入PPARγ的结合腔, 形成了一定的氢键网格, 并且和INT131的结合模式相似。化合物15保留了INT131的U型构象, 但只有磺酰胺键的氮与Tyr327残基的氢键作用保留, 磺酰胺键的氧与Tyr327残基的氢键作用消失, 导致化合物15的激动活性大幅降低, 结合活性与激动活性比值提升, 这佐证了参考文献[10]中报道的Tyr327残基与激动活性相关的推测, 这可能与INT131分子中B片段的改变及A片段中一个氯被三氟甲基取代导致的分子构象的微调有关。

|
Figure 3 Docking of compound INT131 (A) and 15 (B) with 3FUR |
本文基于文献报道的PPARγ部分激动剂INT131的分子结构, 运用电子等排等方法设计并合成了3个骨架结构15个目标化合物。通过初步活性筛选, 发现其中化合物15具有较高的结合活性和很低的激动活性, 可作为PPARγ非激动剂先导物结构进行进一步结构优化, 但是否具有较好的降糖效果和较低的不良反应仍需进一步探究。并通过初步的构效分析, 发现该类化合物中R3=CF3、R4=H时其结合活性与激动活性比值较高, 说明R3=CF3对于提高结合活性与激动活性比值贡献较大; R2是喹啉环时结合活性与激动活性都大幅提高; 并且B片段是吡啶环时, 结合活性与激动活性比值明显提升。喹啉环上氮原子的氧化导致结合活性与激动活性都稍提高, 但其比值降低。这些结果对于该类化合物的进一步结构优化可提供重要参考。
实验部分熔点仪为Yanaco MP-J3显微熔点仪, 温度未校正。核磁共振仪为Varian Mercury 400M, 内标为TMS。质谱仪为Agilent Technologies LC/MS TOF或Thermo Exactive plus-orbitrap。柱色谱分离用粗硅胶(200~300目), 薄层色谱用硅胶GF254购自青岛海洋化工厂。实验所用试剂均为分析纯, 四氢呋喃(THF)、N, N'-二甲基甲酰胺(DMF)和二氯甲烷(DCM)均经过无水处理, 其他溶剂未特别指出则未经处理。
1 化学部分 1.1 a步合成通法将不同取代的硝基苯(2 mmol)、3-羟基喹啉或者3-羟基异喹啉(2.4 mmol)、Cs2CO3 (2.4 mmol)加入到100 mL单口瓶, 加入40 mL DMF或者THF, 35 ℃下搅拌5 h, TLC监测原料完全反应。加适量乙酸乙酯稀释, 水洗2~3次, 饱和氯化钠水溶液洗, 无水硫酸钠干燥。过滤, 浓缩, 柱色谱纯化得中间体。
1.2 b步合成通法将a步产物(2.4 mmol)溶于混合溶液(甲醇-水=20:1), 加热到45 ℃, 搅拌下加入Zn粉(24 mmol)、氯化铵(24 mmol), 温度升至70 ℃, 反应30 min, TLC监测反应完成, 降至室温, 硅藻土过滤, 母液浓缩, 加适量水, 乙酸乙酯萃取两次, 有机相用饱和氯化钠水溶液洗, 然后用无水硫酸钠干燥。过滤, 浓缩所得固体直接用于下一步反应。
1.3 c步合成通法将b步产物(1.1 mmol)溶于无水二氯甲烷, 搅拌下加入吡啶(1 mL)、催化量的DMAP和2, 4-二氯苯磺酰氯(1.2 mmol), 溶液变为鲜红色, 室温搅拌过夜, 次日TLC检测显示反应完成。后处理:将反应液蒸干, 加20 mL乙酸乙酯, 饱和碳酸氢钠溶液洗两次, 水洗, 饱和氯化钠水溶液洗, 无水硫酸钠干燥。过滤, 浓缩, 柱色谱纯化得相应的目标化合物。
1.4 d步合成通法将c步产物(1 mmol)溶于氯仿, 加入3-氯过氧苯甲酸(2 mmol), 室温搅拌过夜。TLC监测反应完成后加氯仿稀释, 用饱和碳酸氢钠水溶液洗, 饱和食盐水洗, 无水硫酸钠干燥, 过滤, 浓缩, 重结晶得氮氧产物。
2 生物活性实验PPARγ配体结合活性:在微孔板中先后加入受试化合物、Fluromone荧光基团以及相应的PPARγ抗体, 充分混匀后, 避光孵育数小时, 检测各孔中荧光强度(供体荧光495 nm, 受体荧光520 nm)。计算TR-FRET比值(520 nm荧光值/495 nm荧光值), 对不同浓度下的受试化合物的TR-FRET比值作浓度-比值拟合曲线。
按以下公式计算IC50及Ki值:
| $ {{K}_{i}}=\frac{I{{C}_{50}}}{1+\frac{\left[\text{tracer} \right]}{{{K}_{\text{D}}}}} $ |
其中[tracer]为检测反应中最大TR-FRET值对应的供体浓度, 本实验中设定为5 nmol·L-1; KD为供体荧光的结合常数, 本实验的KD值为2.8 ± 0.8 nmol·L-1; IC50为达到最大TR-FRET值一半时所对应的待测化合物浓度。
PPARγ配体激动活性:将293E细胞转染荧光素酶报告基因(Peak12-6×Gal4UAS-luci)和表达载体(pcDNA3.1-hPPARγLBD-Gal4DB)共转染, 转染24 h后, 分别加入受试化合物和阳性对照化合物, 化合物处理24 h后裂解细胞, 加入荧光素酶底物, 用多功能酶标仪检测荧光素酶的活性。计算测试化合物不同浓度下相比阴性对照的荧光素酶活性倍数(fold of luciferase activity) = Value化合物 / Value阴性对照。以完全激动剂罗格列酮的最大荧光素酶活性值为100%, 计算受测化合物在不同浓度下的标准化荧光素酶活性百分比(% normalized luciferase activity)[12]。
| [1] | Diamant DM, Heine RJ. Thiazolidinediones in type 2 diabetes mellitus[J]. Drugs, 2003, 63: 1373–1405. DOI:10.2165/00003495-200363130-00004 |
| [2] | DeFronzo RA, Ferrannini E, Groop L, et al. Type 2 diabetes mellitus[J]. Nat Rev Dis Primers, 2015, 1: 1–22. |
| [3] | Feldman PL, Lambert MH, Henke BR. PPAR modulators and PPAR pan agonists for metabolic diseases:the next generation of drugs targeting peroxisome proliferator-activated receptors?[J]. Curr Top Med Chem, 2008, 8: 728–749. DOI:10.2174/156802608784535084 |
| [4] | Rudnicki M, Tripodi GL, Ferrer R, et al. New thiazolidinediones affect endothelial cell activation and angiogenesis[J]. Eur J Pharmacol, 2016, 782: 98–106. DOI:10.1016/j.ejphar.2016.04.038 |
| [5] | Jones D. Potential remains for PPAR-targeted drugs[J]. Nat Rev Drug Discov, 2010, 9: 668–669. DOI:10.1038/nrd3271 |
| [6] | Cain C, Writer S. PPARγ:none is more[J]. Science-Business eXchange, 2011, 4: 1–2. |
| [7] | Han E, Jang SY, Kim G, et al. Rosiglitazone use and the risk of bladder cancer in patients with type 2 diabetes[J]. Medicine, 2016, 95: e2786. DOI:10.1097/MD.0000000000002786 |
| [8] | Ahmadian M, Suh JM, Hah N, et al. PPARγ signaling and metabolism:the good, the bad and the future[J]. Nat Med, 2013, 19: 557–566. |
| [9] | Taygerly JP, Mcgee LR, Rubenstein SM, et al. Discovery of INT131:a selective PPARγ modulator that enhances insulin sensitivity[J]. Bioorg Med Chem, 2013, 21: 979–992. DOI:10.1016/j.bmc.2012.11.058 |
| [10] | Wang MZ, Weiszmann J. INT131:a selective modulator of PPARγ[J]. J Mol Biol, 2009, 386: 1301–1311. DOI:10.1016/j.jmb.2009.01.025 |
| [11] | Lee DH, Huang H, Choi K, et al. Selective PPARγ modulator INT131 normalizes insulin signaling defects and improves bone mass in diet-induced obese mice[J]. Am J Physiol Endocrinol Metab, 2012, 302: E552–60. DOI:10.1152/ajpendo.00569.2011 |
| [12] | Huan Y, Peng J, Wang Y, et al. Establishment and application of screening methods for non-agonist PPARγ ligand[J]. Acta Pharm Sin (药学学报), 2014, 49: 1658–1664. |
| [13] | Garcia-Vallvé S, Guasch L, Tomas-Hernández S, et al. Peroxisome proliferator-activated receptor γ (PPARγ) and ligand choreography:newcomers take the stage[J]. J Med Chem, 2015, 58: 5381–5394. DOI:10.1021/jm501155f |