 2017, Vol. 52
2017, Vol. 52



结核病(tuberculosis, TB)是由结核分支杆菌(Mycobacterium tuberculosis, Mtb)引起的慢性传染性疾病。世界卫生组织2015年《全球结核报告》显示2014年全球有960万人感染结核, 其中印度、印度尼西亚和中国分别占比23%、10%和10%; 150万人死于结核, 其中40万人同时患有艾滋病。结核病与艾滋病已经成为全球死亡率最高的疾病。世卫组织预测, 2014年3.3%新的TB患者和20%接受过治疗的TB患者患有耐多药结核病(multi-drug resistant tuberculosis, MDR-TB), 全球大概有30万的MDR-TB患者, 其中54%来自印度、中国和俄罗斯[1]; MDR-TB患者中又有大概9.7%患有广泛耐药结核病(extensively drug resistant tuberculosis, XDR-TB)[1]。
目前TB的标准治疗方法是异烟肼、利福平、吡嗪酰胺和乙胺丁醇四药联合使用, 每日口服, 持续治疗6~9个月[1]。如果患者严格遵守治疗法则, 这种治疗方法对药敏型(drug-sensitive) TB的治愈率将达到85%~90%[2]。然而, 这4种药物均有较为严重的不良反应, 包括胃肠道炎症或疼痛、肝脏毒性等[3]。由于较长的治疗时间以及严重的不良反应, 病人对这种治疗方法依从性较差, 导致接受治疗的患者中会有20%~30%产生耐药性[4]。MDR-TB患者对异烟肼和利福平有耐药性, 只能使用二线抗TB药物治疗, 治疗时间长达18~20个月, 且不良反应更为严重[5]。而XDR-TB患者不但对异烟肼和利福平产生耐药性, 而且对氟喹诺酮类药物中的一种以及卷曲霉素、卡那霉素和丁胺卡那霉素中的一种或者一种以上产生耐药性, 目前几乎没有有效的治疗方法。考虑到TB患者对现存抗TB药物的较高耐药性, 发现与开发具有新的作用靶标和作用机制的抗TB药物是研究者亟需解决的问题[6]。
Mtb对普通抗生素具很强抗药性, 这与其复杂的细胞壁结构有关。Mtb细胞壁组成成分及合成途径是结核杆菌特有的, 在哺乳动物细胞中完全不存在, 因此, 阻碍细胞壁形成是目前开发抗TB药物的主要思路之一。
分枝杆菌膜蛋白3 (mycobacterial membrane protein large 3, MmpL 3) 在构建细胞壁必须成分的转运过程中起到关键作用(本文第三部分中将详细介绍), 因此筛选与设计抑制MmpL3靶标的小分子化合物成为近年来开发抗TB药物的热点之一。近年来, MmpL3已经成为最成功的抗TB药物靶标。基于全细胞的高通量筛选已经确证7类化学骨架不同的MmpL3抑制剂。本文将简要论述这几类MmpL3抑制剂, 介绍靶标确证方法, 并进一步探讨MmpL3抑制剂的作用机制。
1 研发中的MmpL3抑制剂 1.1 乙二胺类抑制剂在利用组合化学固相合成方法合成一系列乙胺丁醇类似物以进一步研究其构效关系的过程中, Barry团队发现了化合物SQ109 (图 1)[7]。SQ109作用于MmpL3转运蛋白, 抑制分枝菌酸的转运, 进而产生抗TB活性[8], 目前已完成二期临床研究。

|
Figure 1 The structures of SQ109 and its analogues |
体外活性测试中, SQ109对敏感型和耐药型Mtb的MIC值分别为0.6~1 μmol·L−1和0.6~2 μmol·L−1。体外及体内活性测试结果表明, SQ109的活性均要明显优于乙胺丁醇[9]。此外, 对于巨噬细胞中的Mtb, SQ109的活性与异烟肼相当, 明显优于乙胺丁醇。体外联合用药结果显示SQ109与已上市或在研抗TB药物具有协同作用。同时, SQ109显示杰出的药代性质, 在犬、大鼠和小鼠中的口服生物利用度分别为2.4%~5%、12%和3.8%;尽管其口服生物利用度较低, 但组织分布研究显示其在小鼠的肺和脾部位的浓度达到MIC值的45倍。在一个8周的小鼠模型中, SQ109代替乙胺丁醇进行四药联用, 疗效得到显著提高。此外, 当SQ109联合其他标准抗TB药物或者贝达喹啉使用时, 其治疗效果也要优于标准治疗方案[10]。临床Ⅰ期研究显示SQ109在人体内单一剂量或多剂量使用时均具有良好的安全性及耐受性。Ⅱ期临床研究表明SQ109单一疗法每日剂量为75、150和300 mg或者和标准剂量的利福平联合使用时均呈现出良好的安全性及耐受性[9]。
SQ609 (图 1)在体外具有很好的抗TB活性。20天Mtb感染引起的体重损失的小鼠模型实验中, SQ609可以很好地防止小鼠体重的损失并提高其生存能力。停止给药后, SQ609的治疗效果可延长10~15天左右[11]。
Eric Oldfiel团队合成了11个新的类似物, 其中化合物3和4 (图 1)的MIC值分别为0.02~0.05和0.05 μmol·L−1, 体外活性优于SQ109。结果表明根据生物电子等排原理, 用氧原子代替NH后也可维持化合物的抗TB活性[12]。
对该类化合物现有研究工作结果初步分析, 构效关系如下: ① 乙二胺骨架是抗TB活性的结构基础; ② 乙二胺骨架上取代烷基的体积及性质对活性有重要的影响; ③ 结构中两个碱性NH不是活性所必需的; ④ 长链烷基与大体积烷基有利于提高活性(图 2)。

|
Figure 2 SAR analysis of SQ109 and its analogues |
Franzblau团队[13]在对6 800个化合物进行表型筛选时发现了苗头化合物5 (表 1), 它对Mtb有较强的抗菌活性, MIC值为0.93 μmol·L−1。然而化合物5在pH 6.8时溶解度小于4 μmol·L−1, 其脂水分布系数cLogP为4.48, 且在小鼠肝微粒体中清除率较高(Clint=693 μL·min−1·mg−1)。随后Kozikowski团队[14]经过优化, 确定了先导化合物6 (表 1)。先导物6对MtbH37Rv的MIC值为0.013 μmol·L−1, 对Vero细胞的IC50值为54 μmol·L−1, 表明其具有较高的安全选择性。
| Table 1 The structures of indole-2-carboxamide inhibitors |
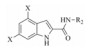

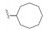

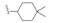
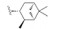
Novarits公司研究团队对苗头化合物5的结构优化确定了两个临床前候选药物NITD-304和NITD-349 (表 1)。NITD-304和NITD-349对H37Rv菌株显示良好活性, MIC值分别为0.015和0.023 μmol·L−1, 对耐药性菌株MIC值在0.02~0.08 μmol·L−1。体外实验中, NITD-304和NITD-349在小鼠和人肝微粒体中具有适中的清除率。进行口服药代性质评估时, 两个候选药物在不同动物属种均呈现良好药代性质:前者口服生物利用度在15%~76%, 而后者口服生物利用度在38%~100%。体内活性测试实验中, 给药剂量为25和75 mg·kg−1时, 前者分别降低2.6和4.8个菌落形成单位(colony-forming units, CFU), 后者分别降低2.4和4.6个logCFU[15]。
2016年Kozikowski团队[16]又确定一个新的先导物9 (表 1), 对H37Rv的MIC值达到0.012 μmol·L−1, 对Vero细胞的IC50值大于192 μmol·L−1, 呈现出很高的安全选择性。
对吲哚-2-甲酰胺类抑制剂现阶段研究工作的结果综合分析, 可将构效关系归纳如下: ① 吲哚-2-甲酰胺化学骨架是其活性基础; ② 吲哚环上的NH以及酰胺键上的NH对活性有重要影响; ③ 4, 6-二氯取代或4, 6-二氟取代可提高其代谢稳定性; ④ 抗TB活性与cLogP之间呈正相关; ⑤ 极性基团取代可提高化合物的溶解性, 但同时也会降低其抗TB活性(图 3)。

|
Figure 3 SAR analysis of indole-2-carboxamideinhibitors |
Lee团队[17]利用微量肉汤稀释法对包含约12 000个化合物的库进行筛选, 确定了1-金刚烷基-3-苯基脲类抑制剂。这类化合物的作用靶标也是MmpL3, 同时对人体可溶环氧化物水解酶(human soluble epoxide hydrolase, hsEH)以及Mtb环氧化物水解酶具有抑制作用。AU1235 (表 2)是其中的一个代表性的苗头化合物, 它对Mtb的MIC达到0.3 μmol·L−1。
| Table 2 The structures of adamantly urea inhibitors |


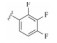
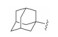
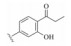

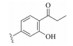
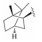
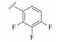
然而, AU1235这系列化合物的溶解性与体外药代性质较差。随后, 在对包含有1 600个脲类化合物的化合物库进行筛选时发现1-金刚烷基-3-苯基脲的对位有极性基团时可维持活性, 同时发现化合物11口服给药后在小鼠血浆中的浓度高于其MIC, 然而其并未消除小鼠模型中Mtb的感染数量。体内活性的丧失究竟是由高蛋白结合率导致或者其他原因所致还有待进一步实验证明[18]。基于生物电子等排原理, 以吡啶环、嘧啶环、三嗪环、噁唑环等芳杂环代替原先的苯环, 合成一系列1-金刚烷基-3-芳杂环脲类化合物, 在维持抗TB活性的同时体外药代性质得到改善, 选择性也有所提高, 但这些化合物并不具有体内活性[12, 19]。
最近, 中国学者宋宏锐的团队[20]为增强AU1235与靶点的相互作用, 将金刚烷部分改造为其他类取代基, 设计合成一系列脲类衍生物, 其中化合物13具有很好的抗TB活性(MIC=0.060 μg·mL−1), 并且细胞毒性和脂水分配系数均较低, 值得进一步研究。
此类抑制剂的初步构效关系综述如下: ① 1-环烷基-3-苯基双取代脲母核是其抗TB活性所必需; ② 对于环烷基部位, 大位阻基团如金刚烷基等活性更高; ③ 苯环上可容纳各种不同的取代基(图 4)。

|
Figure 4 SAR analysis of adamantly urea inhibitors |
在利用表型高通量筛选化合物的过程中, 不同的研究团体均各自独立的确证了THPP这类化学骨架的MmpL3抑制剂。
葛兰素史克公司(GSK)的Cammack团队发现苗头化合物14 (表 3), 其对H37Rv菌株的MIC达到0.3 μmol·L−1, 但在多种溶剂中的溶解度都很差, 无法进行体内药效研究。随后的结构优化过程发现化合物15 (表 3), 其维持体外抗TB活性(对H37Rv的MIC是0.16 μmol·L−1)的同时, 能够在一次口服剂量50 mg·kg−1的条件下达到可接受的血浆浓度(AUC24h=26.5 μg·h·mL−1)。对小鼠给药100和300 mg·kg−1后, 肺部感染菌数分别降低1.5和2.2 logCFU。然而化合物15结构中包含苯并二噁茂环片段, 可能存在与其他药物相互作用及可能的毒性等问题[21]。
| Table 3 The structures of THPP inhibitors of GSK |


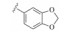
Novarits公司的Jiricek团队[22]也合成了系列THPP骨架的抑制剂(表 4), 其中NITD-192活性最好, 但它具有较高的脂水分配系数(cLogP=6.3) 和血浆蛋白结合率(plasma protein binding, PPB > 99.0%)以及低溶解度, 成药性需改进。随后Novarits的研究人员改善NITD-192的类药性质, 尽管获得的新类似物如化合物17~19等具有较好的体内药代性质, 却完全丧失体内活性; 而NITD-192在给药剂量为100 mg·kg−1时具有和乙胺丁醇相当的体内活性, 并对所有的MDR-TB菌株有抑制活性, 因此NITD-192在治疗MDR-TB方面仍是很有前景的候选药物[12, 23]。总结GSK和Novarits的工作: ① 四氢嘧啶环上的NH、吡唑环以及二级酰胺侧链是抗TB活性所必须; ② (5R, 7S)构型的骨架具有抗TB活性; ③ 两侧苯环上引入极性基团会导致活性降低甚至丧失; ④ 化合物脂水分布系数降低会导致其体内活性降低(图 5)。
| Table 4 Structures of THPP inhibitors of Novarits |

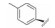




|
Figure 5 SAR analysis of THPP inhibitors |
研究者通过基于全细胞表型筛选确证了1, 5-二苯基吡咯类化学骨架的MmpL3抑制剂, 并发现了BM212[19]、BM521[24]、BM533[25]和BM579[25]这4个活性化合物(表 5)。与母体化合物BM212相比, BM521、BM533和BM579具有很好的抗TB活性(MIC值在0.16~0.20 μmol·L−1), 但类药性评估结果显示这3个活性化合物在小鼠肝微粒体中具有较高的清除率。其结构中硫代吗啉部分是在微粒体内不稳定的原因, 故Biava团队[23]集中精力修饰这一结构部位, 用吗啉环代替硫代吗啉环, 合成了系列化合物, 并将BM635 (表 5)确定为候选化合物。
| Table 5 The structures of 1, 5-diphenyl pyrrole inhibitors |

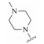

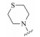
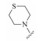

BM635具有很好的体外抗TB活性(MIC值0.12 μmol·L−1)及小鼠微粒体稳定性(1.4 mL·min−1·g−1), 并且Tox50高于MIC值100多倍。基于其优良的类药性质, Biava团队[23]进一步研究其体内药代动力学性质及其体内活性, 在小鼠急性感染模型中, BM635呈现出很好的抗TB活性, ED99值[使肺部CFU (菌落形成单位)降低99%的有效剂量值]为49 mg·kg−1; 给药剂量为50 mg·kg−1时, 体内半衰期为1 h, 体内最高浓度达到1.62 μmol·L−1。综合考量AUC与ED99值, BM635体内每单位有效浓度的效能与一线抗TB药物异烟肼、利福平、莫西沙星相比具较强优势, 故对其进一步的结构优化将为抗TB提供具有更高活性及更优良类药性质的候选药物。1, 5-二苯基吡咯类抑制剂的初步构效关系可归纳如下: ① 1, 5-二苯基吡咯骨架是其抗TB活性的结构基础; ② 两个苯环对位取代抗TB活性更好; ③ F对位取代N-1位苯环可增加其活性并降低细胞毒性; ④ 烷基对位取代C-5位苯环可增加其活性; ⑤ C-2位侧链长度增加会导致活性丧失; ⑥ C-3位的吗啉环可增加微粒体稳定性(图 6)。

|
Figure 6 SAR analysis of 1, 5-diphenyl pyrrole inhibitors |
GSK的Barturen团队[26]通过高通量表型筛选确定具有螺环化学骨架的苗头化合物GSK2200150A (表 6)。尽管其对H37Rv的MIC是0.3 μmol·L−1, 但物理化学性质及体外药代动力学参数均不理想, 因此无法进行体内药效研究。保留其N-亚甲基苯基结构以维持其抗TB活性的同时, 研究着重于优化其物化性质以改善类药性质, 最终获得化合物26 (表 6)。
| Table 6 The structures of spirocycle inhibitors |

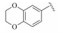
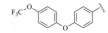
尽管化合物26具有较高的脂水分布系数, 但其体外活性显著提高(H37Rv MIC 0.06 μmol·L−1), 且一次口服给药50 mg·kg−1后, 血浆内的暴露程度达到了可接受的程度, 因此研究人员进一步对其进行小鼠急性感染Mtb模型实验。结果显示, 给药50和100 mg·kg−1后, 小鼠肺部的Mtb分别减少了2.3和2.7个logCFU, 与Ⅲ期临床的抗TB候选药物莫西沙星相比在给药剂量方面具有一定优势[19, 26]。
1.7 苯并咪唑类抑制剂在对布罗德研究所的2万个小分子化合物库进行全细胞表型筛选的过程中Hung团队[27]确证了化合物C215 (图 7)的抗结核作用。

|
Figure 7 The structure of C215 |
C215对结核分枝杆菌的抑制90%细菌繁殖的浓度(IC90)为16 μmol·L−1, 在巨噬细胞中C215的细菌效能增强, 同时其对哺乳动物细胞的毒性较小, IC90为37.5 μmol·L−1[28]。
2 靶标确证针对上述几类MmpL3抑制剂的作用机制研究, MmpL3作为TMM转运蛋白的功能逐渐被发现与揭示。
Mtb的细胞壁含有丰富的脂质结构, 使得分支杆菌可以存活于严酷的环境并抵抗宿主的免疫反应[29]; 其核心层霉菌酸−阿拉伯半乳聚糖−肽聚糖复合物(mycolic acid-arabinogalactan-peptidoglycan complex, MAPc)[30]对维持细胞的活性有重要作用[31] (图 8); 研究还表明霉菌酸与Mtb的毒性有关[32]。霉菌酸的生物合成途径较为复杂, 由多功能的Ⅰ型脂肪酸合成酶(FAS-Ⅰ)和多酶复合物的Ⅱ型脂肪酸合成酶(FAS-Ⅱ)共同完成[33, 34]。霉菌酸可以键合到海藻糖二糖的6-羟基上形成TMM[35]。TMM是海藻糖二霉菌酸酯(trehalosedimycolate, TDM)和其他细胞壁上的霉菌酸酯的前体化合物。

|
Figure 8 Schematic representation of the mycobacterial cell wall |
TMM从胞浆内转运到周质边缘需要跨膜蛋白MmpL3的参与[36, 37]。MmpL3属于分枝杆菌膜蛋白大家族, MmpL蛋白则属于抗瘤细胞分裂(resistance-nodulation-cell division, RND)蛋白超家族[38]。Mtb中有13种RND蛋白, 即13种MmpL蛋白, 分子质量在100~122 kDa之间[29]。研究发现Mtb编码的13种MmpL蛋白中只有MmpL3是维持Mtb活性所必需的, 它调节TMM的跨膜转运。因此, 抑制MmpL3靶标可以阻断TMM的跨膜转运, 从而阻碍Mtb细胞壁的形成, 达到抑制Mtb的目的。
在对上述几类化合物的耐药突变菌株进行全基因测序[39]时发现mmpL3基因发生点突变。自发的耐药菌株在接受药物治疗时趋向于在MmpL3蛋白处发生突变以此缓解抑制剂的早期毒性效应。这些突变株均显示了中等程度的耐药性: BM212、AUs、SQ109、吲哚-2-甲酰胺类抑制剂和THPPs的MIC值分别增加了16~33倍、8倍、4~8倍、4~122倍和8~70倍[40]。
用1x、5x和10x MIC浓度的上述几类抑制剂处理Mtb时, 尽管细胞内霉菌酸酯的总量没有发生变化, 但是霉菌酸酯的亚细胞分布发生变化。上述几类化合物处理的Mtb细胞中, TDM与嵌合到细胞壁阿拉伯半乳聚糖上的霉菌酸酯的数量都有明显下降, 同时可以观察到细胞内TDM前体TMM数量的增加。这说明上述化合物并不抑制霉菌酸酯的生物合成, 而是抑制TMM的转运过程。最近研究提出:上述几类化合物抑制TMM的转运是通过一个间接的过程。BM212、THPPs、吲哚-2-甲酰胺类[41]、AUs[42]和SQ109影响跨膜质子浓度(ΔpH)和膜电位(ΔΨ)中一种或者两种, 从而驱散PMF, 进而抑制MmpL3调节的转运过程[10]。SQ109不但作为解偶联剂驱散质子泵, 同时抑制涉及维生素K2和ATP合成的酶[43] (图 9)。但为何自发的耐药突变映射到mmpL基因, 这一问题仍没有答案, 有待进一步的研究加以揭示。

|
Figure 9 MOA of SQ109 |
2012年FDA批准了近40年来的首个抗TB新药贝达喹啉用于治疗成人多重耐药结核, 这标志着人类在与MDR-TB及XDR-TB的抗争中迈出了重要的一步, 但目前的形势依然严峻。
MmpL3靶标是近年来最为成功的抗TB靶标之一, 多方的研究成果提供了一系列化学骨架不同的候选药物。这些MmpL3抑制剂不但化学结构新颖, 同时具有完全不同于传统抗TB药物的作用机制, 对DS-TB、MDR-TB、XDR-TB均呈现出很好的活性; 但现有化合物同时也存在着诸如脂水分配系数较高(clogP > 4.2)、溶解度较低等药代性质方面的缺陷。目前还没有任何研究团队解析出MmpL3蛋白的完整晶体结构, 这给基于理性的方法设计MmpL3抑制剂带来了一定的困难。
未来的研究取决于结构生物学的突破, 如能解析出MmpL3蛋白结构, 药化工作者可运用更多的技术手段合理地对先导物进行结构修饰, 改善现有的MmpL3抑制剂的药代性质, 有望促进该类抑制剂的早日上市, 为TB患者带来更多、更好的用药选择。
| [1] | Global tuberculosis report 2015[M]. 20th ed. World Health Organization, 2015:1-3. |
| [2] | Cooper CB. Development of Mycobacterium tuberculosis whole cell screening hits as potential antituberculosis agents:miniperspectives series on phenotypic screening for antiinfective targets[J]. J Med Chem, 2013, 56: 7755–7760. DOI:10.1021/jm400381v |
| [3] | Quy H, Lönnroth K, Lan N, et al. Treatment results among tuberculosis patients treated by private lung specialists involved in a public-private mix project in Vietnam[J]. Int J Tuberc Lung Dis, 2003, 7: 1139–1146. |
| [4] | Goldberg DE, Siliciano RF, Jacobs WR. Outwitting evolution:fighting drug-resistant TB, malaria, and HIV[J]. Cell, 2012, 148: 1271–1283. DOI:10.1016/j.cell.2012.02.021 |
| [5] | Zumla A, Abubakar I, Raviglione M, et al. Drug-resistant tuberculosis-current dilemmas, unanswered questions, challenges, and priority needs[J]. J Infect Dis, 2012, 205(suppl 2): S228–S240. |
| [6] | Rivers EC, Mancera RL. New anti-tuberculosis drugs in clinical trials with novel mechanisms of action[J]. Drug Discov Today, 2008, 13: 1090–1098. DOI:10.1016/j.drudis.2008.09.004 |
| [7] | Lee RE, Protopopova M, Crooks E, et al. Combinatorial lead optimization of[1, 2] -diamines based on ethambutol as potential antituberculosis preclinical candidates[J]. J Comb Chem, 2003, 5: 172–187. DOI:10.1021/cc020071p |
| [8] | Tahlan K, Wilson R, Kastrinsky DB, et al. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis[J]. Antimicrob Agents Chem, 2012, 56: 1797–1809. DOI:10.1128/AAC.05708-11 |
| [9] | Sacksteder KA, Protopopova M, Barry CE, et al. Discovery and development of SQ109:a new antitubercular drug with a novel mechanism of action[J]. Future Microbiol, 2012, 7: 823–837. DOI:10.2217/fmb.12.56 |
| [10] | Poce G, Consalvi S, Biava M. MmpL3 inhibitors:diverse chemical scaffolds inhibit the same target[J]. Mini Rev Med Chem, 2016, 16: 1274–1283. DOI:10.2174/1389557516666160118105319 |
| [11] | Bogatcheva E, Hanrahan C, Nikonenko B, et al. Identification of SQ609 as a lead compound from a library of dipiperidines[J]. Bioorg Med Chem Lett, 2011, 21: 5353–5357. DOI:10.1016/j.bmcl.2011.07.015 |
| [12] | Li K, Schurig-Briccio LA, Feng X, et al. Multitarget drug discovery for tuberculosis and other infectious diseases[J]. J Med Chem, 2014, 57: 3126–3139. DOI:10.1021/jm500131s |
| [13] | Collins L, Franzblau SG. Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium[J]. Antimicrob Agents Chem, 1997, 41: 1004–1009. |
| [14] | Onajole OK, Pieroni M, Tipparaju SK, et al. Preliminary structure-activity relationships and biological evaluation of novel antitubercular indolecarboxamide derivatives against drugsusceptible and drug-resistant Mycobacterium tuberculosis strains[J]. J Med Chem, 2013, 56: 4093–4103. DOI:10.1021/jm4003878 |
| [15] | Kondreddi RR, Jiricek J, Rao SP, et al. Design, synthesis, and biological evaluation of indole-2-carboxamides:a promising class of antituberculosis agents[J]. J Med Chem, 2013, 56: 8849–8859. DOI:10.1021/jm4012774 |
| [16] | Stec J, Onajole OK, Lun S, et al. Indole-2-carboxamidebased MmpL3 inhibitors show exceptional antitubercular activity in an animal model of tuberculosis infection[J]. J Med Chem, 2016, 59: 6232–6247. DOI:10.1021/acs.jmedchem.6b00415 |
| [17] | Brown JR, North EJ, Hurdle JG, et al. The structure activity relationship of urea derivatives as anti-tuberculosis agents[J]. Bioorg Med Chem, 2011, 19: 5585–5595. DOI:10.1016/j.bmc.2011.07.034 |
| [18] | Scherman MS, North EJ, Jones V, et al. Screening a library of 1600 adamantyl ureas for anti-Mycobacterium tuberculosis activity in vitro and for better physical chemical properties for bioavailability[J]. Bioorg Med Chem, 2012, 20: 3255–3262. DOI:10.1016/j.bmc.2012.03.058 |
| [19] | Deidda D, Lampis G, Fioravanti R, et al. Bactericidal activities of the pyrrole derivative BM212 against multidrugresistant and intramacrophagic Mycobacterium tuberculosis strains[J]. Antimicrob Agents Chem, 1998, 42: 3035–3037. |
| [20] | Wang H, Wen H, Cui HQ, et al. Synthesis and anti-tubercular activity of 1-(2-adamantyl)-3-(2, 3, 4-trifluorophenyl)urea derivatives[J]. Chin J Med Chem(中国药物化学杂志), 2016, 26: 280–287. |
| [21] | Remuiñán MJ, Pérez-Herrán E, Rullás J, et al. Tetrahydropyrazolo[1, 5-a] pyrimidine-3-carboxamide and N-benzyl-6', 7'-dihydrospiro[piperidine-4, 4'-thieno[3, 2-c]pyran] analogues with bactericidal efficacy against Mycobacterium tuberculosis targeting MmpL3[J]. PLoS One, 2013, 8: e60933. DOI:10.1371/journal.pone.0060933 |
| [22] | Yokokawa F, Wang G, Chan WL, et al. Discovery of tetrahydropyrazolopyrimidine carboxamide derivatives as potent and orally active antitubercular agents[J]. ACS Med Chem Lett, 2013, 4: 451–455. DOI:10.1021/ml400071a |
| [23] | Poce G, Bates RH, Alfonso S, et al. Improved BM212 MmpL3 inhibitor analogue shows efficacy in acute murine model of tuberculosis infection[J]. PLoS One, 2013, 8: e56980. DOI:10.1371/journal.pone.0056980 |
| [24] | Biava M, Porretta GC, Poce G, et al. Antimycobacterial agents. Novel diarylpyrrole derivatives of BM212 endowed with high activity toward Mycobacterium tuberculosis and low cytotoxicity[J]. J Med Chem, 2006, 49: 4946–4952. DOI:10.1021/jm0602662 |
| [25] | Biava M, Porretta GC, Poce G, et al. Identification of a novel pyrrole derivative endowed with antimycobacterial activity and protection index comparable to that of the current antitubercular drugs streptomycin and rifampin[J]. Bioorg Med Chem, 2010, 18: 8076–8084. DOI:10.1016/j.bmc.2010.09.006 |
| [26] | Rullas J, García JI, Beltrán M, et al. Fast standardized therapeutic-efficacy assay for drug discovery against tuberculosis[J]. Antimicrob Agents Chem, 2010, 54: 2262–2264. DOI:10.1128/AAC.01423-09 |
| [27] | Stanley SA, Grant SS, Kawate T, et al. Identification of novel inhibitors of M. tuberculosisgrowth using whole cell based high-throughput screening[J]. ACS Chem Biol, 2012, 7: 1377–1384. DOI:10.1021/cb300151m |
| [28] | Stanley SA, Grant SS, Kawate T, et al. Identification of novel inhibitors of M. tuberculosis growth using whole cell based high-throughput screening[J]. ACS Chem Biol, 2012, 7: 1377–1384. DOI:10.1021/cb300151m |
| [29] | Rayasam GV. MmpL3 a potential new target for development of novel anti-tuberculosis drugs[J]. Expert Opin Ther Target, 2014, 18: 247–256. DOI:10.1517/14728222.2014.859677 |
| [30] | Yamaryo-Botte Y, Rainczuk AK, Lea-Smith DJ, et al. Acetylation of trehalose mycolates is required for efficient MmpLmediated membrane transport in corynebacterineae[J]. ACS Chem Biol, 2014, 10: 734–746. |
| [31] | Kaur D, Guerin ME, Škovierová H, et al. Biogenesis of the cell wall and other glycoconjugates of Mycobacterium tuberculosis[J]. Adv Appl Microbiol, 2009, 69: 23–78. DOI:10.1016/S0065-2164(09)69002-X |
| [32] | Vander Beken S, Al Dulayymi J a R, Naessens T, et al. Molecular structure of the Mycobacterium tuberculosis virulence factor, mycolic acid, determines the elicited inflamematory pattern[J]. Eur J Immunol, 2011, 41: 450–460. DOI:10.1002/eji.201040719 |
| [33] | Schweizer E, Hofmann J. Microbial type Ⅰ fatty acid synthases (FAS):major players in a network of cellular FAS systems[J]. Microbiol Mol Biol Rev, 2004, 68: 501–517. DOI:10.1128/MMBR.68.3.501-517.2004 |
| [34] | Nataraj V, Varela C, Javid A, et al. Mycolic acids:deciphering and targeting the Achilles' heel of the tubercle bacillus[J]. Mol Microbiol, 2015, 98: 7–16. DOI:10.1111/mmi.13101 |
| [35] | Nobre A, Alarico S, Maranha A, et al. The molecular biology of mycobacterial trehalose in the quest for advanced tuberculosis therapies[J]. Microbiology, 2014, 160: 1547–1570. DOI:10.1099/mic.0.075895-0 |
| [36] | Varela C, Rittmann D, Singh A, et al. MmpL genes are associated with mycolic acid metabolism in mycobacteria and corynebacteria[J]. Chem Biol, 2012, 19: 498–506. DOI:10.1016/j.chembiol.2012.03.006 |
| [37] | Belardinelli JM, Yazidi A, Yang L, et al. Structure-function profile of MmpL3, the essential mycolic acid transporter from Mycobacterium tuberculosis[J]. ACS Infect Dis, 2016, 2: 702–713. DOI:10.1021/acsinfecdis.6b00095 |
| [38] | Domenech P, Reed MB, Barry CE. Contribution of the Mycobacterium tuberculosis MmpL protein family to virulence and drug resistance[J]. Infect Immun, 2005, 73: 3492–3501. DOI:10.1128/IAI.73.6.3492-3501.2005 |
| [39] | Goldman RC. Why are membrane targets discovered by phenotypic screens and genome sequencing in Mycobacterium tuberculosis?[J]. Tuberculosis, 2013, 93: 569–588. DOI:10.1016/j.tube.2013.09.003 |
| [40] | Li W, Upadhyay A, Fontes FL, et al. Novel insights into the mechanism of inhibition of MmpL3, a target of multiple pharmacophores in Mycobacterium tuberculosis[J]. Antimicrob Agents Chem, 2014, 58: 6413–6423. DOI:10.1128/AAC.03229-14 |
| [41] | Lun S, Guo H, Onajole OK, et al. Indoleamides are active against drug-resistant Mycobacterium tuberculosis[J]. Nat Commun, 2013, 4: 2907. |
| [42] | Grzegorzewicz AE, Pham H, Gundi VA, et al. Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane[J]. Nat Chem Biol, 2012, 8: 334–341. DOI:10.1038/nchembio.794 |
| [43] | Foss MH, Pou S, Davidson PM, et al. Diphenylethermodified 1, 2-diamines with improved drug properties for development against Mycobacterium tuberculosis[J]. ACS Infect Dis, 2016, 2: 500–508. DOI:10.1021/acsinfecdis.6b00052 |