 2017, Vol. 52
2017, Vol. 52

编者按
新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
基于流感神经氨酸酶的晶体结构和生化研究, 以底物的过渡态作为出发点, 将分子模拟的结果指导抑制剂的设计, 在设计合成的有限化合物中, 诞生了首创的扎那米韦。然而也由于过分拘泥模仿底物的结构, 过强的极性导致扎那米韦药代的缺陷和用药的局限性。奥塞米韦也是基于酶的三维结构和模拟底物过渡态, 但注意到成药性的要求, 也在为数不多的化合物中, 成功地合成出结构简单而合理的口服抗流感药。从研发和上市的顺序看, 奥塞米韦是后继药物, 但超越了首创的扎那米韦。
人流感主要由A、B两型流感病毒感染发生, A型病毒又可根据包膜上的两种糖蛋白的抗原特性分成亚型, 这两种糖蛋白称作血凝素和神经氨酸酶, 血凝素有16种(H1~H16), 神经氨酸酶有9种(N1~N9), 近年发生的流感大流行是由H5N1和H1N1感染。流感病毒极容易变异。
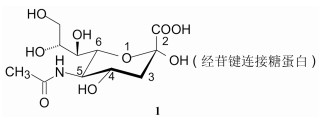
1.1 血凝素和神经氨酸酶血凝素(hemagglutinin, HA)和神经氨酸酶(neuroaminidase, NA, 又称唾液酸酶sialidase)是两种酶, 它们识别并结合的底物是N-乙酰神经氨酸(1, N-acetylneuraminic acid, 又称唾液酸, sialic acid)片段, 后者是人上呼吸道上皮细胞膜上的糖缀合物的末端糖结构, 血凝素结合了唾液酸片段, 将病毒带到细胞膜上, 经病毒包膜与细胞膜融合使病毒进入细胞内(内化)。病毒神经氨酸酶的作用是切掉唾液酸与糖缀合物连接的糖苷键, 将子代的病毒颗粒释放到上皮细胞中, 完成了播散周期。抑制神经氨酸酶以阻止糖苷的裂解, 导致病毒颗粒持续结合于血凝素上, 可阻断病毒的传染。血凝素和神经氨酸酶都是抗流感病毒的靶标, 当今研究较多的是神经氨酸酶。
|
神经氨酸酶是种糖蛋白, 为4个亚基构成的四聚体, 经疏水性的N-端键合于病毒包膜上。其晶体结构于1983年经X-射线衍射解析(Colman PM, Varghese JN, Laver WG. Structure of the catalytic and antigenic sites in influenza virus neuroaminidase. Nature, 1983, 303: 41-44; Varghese JN, McKimm-Breschkin J, Caldwell JB, et al. The structure of the complex between influenza virus neuraminidase and sialic acid, the viral receptor. Proteins, 1992, 14: 327-332)。唾液酸定位于神经氨酸酶的催化中心, 其结合方式是2位羧基与三个精氨酸簇形成静电结合; 5位的乙酰氨基形成两个氢键: NH与水分子结合, C=O与Arg152结合, 甲基结合于Trp178与Ile222构成的疏水腔内; 6位链上的羟基与Glu276的羧基形成二齿氢键, 如图 1所示。

|
图 1 唾液酸与神经氨酸酶的晶体结构图 |
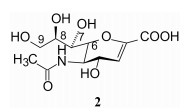
Itzstein等基于神经氨酸酶(apo protein)和酶与唾液酸复合物(complex)的晶体结构, 并通过分子模拟研究, 确定了底物与神经氨酸酶结合特征和基团的能量贡献, 提出了如图 2所示的酶水解唾液酸苷的反应历程。认为底物在游离状态下吡喃糖环呈椅式构象存在, 2位羧基处于直立键, 苷键采取平展位置(2a)。在酶活性中心的氨基酸残基作用下, 带正电荷的Arg371 (以及Arg 118和Arg292) 与带负电荷的2位羧基发生盐键结合, Arg151和Arg152通过与水分子氢键结合, 形成了与苷键的结合网络。这样, 使糖环由椅式变形成船式, 羧基呈假平展键取向, 苷键成为直立键, 并在环中O1原子未偶电子的助力下, 发生成键电荷的重新分布(2b), 形成了苷元即将离去的过渡态, 此时生成的C2正电荷被O1的未偶电子稳定化(p-π共轭), 吡喃环成为半椅式的氧鎓离子, 鎓离子同时被酶的Glu277的负电荷稳定(2c)。水分子形成的OH-向sp2杂化的C2进攻, 恢复了C2的sp3杂化态(2d), 吡喃糖环经过渡态(2e)向产物转变, 从酶的活性中心释放唾液酸, 羧基处于直立键位置(2f), 是由于OH-以SN1机制进攻C2所致, NMR也证明初产物是α异构体, 并逐渐经变旋作用使羧基处于更稳定的平展构型的β体(2g) (Taylor NR, von Itzstein M. Molecular modeling studies on ligand binding to sialidase from influenza virus and the mechanism of catalysis. J Med Chem, 1994, 37: 616-624)。

|
图 2 神经氨酸酶水解唾液酸苷的历程(唾液酸的未变化基团省略) |
该反应机制得到了实验的证明, Burmeister等通过晶体学研究发现流感B病毒的神经氨酸酶催化水解唾液酸苷, 得到了Δ2唾液酸(2, Neu5Ac2en), 2的结构与图 2e的过渡态非常相似, 而且还证明2是神经氨酸酶的抑制剂, 这为设计过渡态类似物提供了有力的根据(Burmeister WP, Henrissat B, Bosso C, et al. Influenza B virus neuraminidase can synthesise its own inhibitor. Structure, 1993, 1: 19-26)。
|
前面提出的水解机制和所涉及的功能残基, 以及过渡态的分子构象变化, 为设计过渡态类似物提供了依据, 接下来需要了解活性中心与底物(或抑制剂)分子的结合模式。为此, 应用了分子模拟的研究方法, 用GRID软件计算并定义活性中心的氨基酸残基与底物的相互作用。为此, 以COO-、NH3+、OH和CH3等4个探针基团扫描活性中心的范德华表面, 分别确定负电荷、正电荷、极性基团和疏水相互作用范围。
2.2.1 羧酸根探针带有负电荷的羧酸根扫描确定出一个相互作用区域, 相当于唾液酸的羧基在活性中心所处的位置, 映射出Arg118、Arg292和Arg371构成的三角区(尤其是Arg371) 的重要性, 也提示抑制剂的设计应有羧基或提供负电荷的基团。
2.2.2 铵离子特征由于神经氨酸酶在pH 5.5呈现最适活性, 碱性的胺类在pH 5.5环境下被质子化, 带有正电荷, 因而用NH3+作为探针, 计算分子表面的能量状态。结果表明有3个区域呈现相互作用:在唾液酸的4-OH附近; 糖环C4的下面; 甘油侧链和5位N-乙酰基之间的区域。
2.2.3 羟基探针羟基探针显示的相互作用与上述的羧酸根的位置重合, 因而未能提供新的相互作用信息。
2.2.4 甲基探针甲基探针发现的疏水性相互作用区域是乙酰胺基侧链上的甲基位置, 对应于Trp178构成的疏水腔。
这些探针勾画的区域, 映射出酶活性部位发生结合作用的环境, 为抑制剂的设计提供了信息和结构背景。
3 设计抑制剂 3.1 模拟过渡态结构根据神经氨酸酶与唾液酸复合物的晶体结构、分子模拟预示活性中心的结合特征, 以及推测的水解反应机制, 抑制剂的设计首先合成了化合物2, 它是唾液酸1的Δ2, 3类似物, 环上的其他取代基未做改变。由于C2和C3为sp2杂化态, 与氧原子的未偶电子对形成p-π共轭, 导致吡喃环变形, 类似于过渡态的氧鎓离子态的平面构象。
化合物2对流感A病毒神经氨酸酶有较弱抑制活性, Ki = 4 μmol·L-1, 分子模拟的结构与实验得到的晶体数据相近。实验还证明将酶活性中心的Arg152突变为Lys, Glu277突变为Asp, 化合物2对发生突变的酶失去抑制活性, 推测是由于Lys和Asp残基链变短, 达不到参与结合位置, 从而佐证了Arg152和Glu277参与同2的结合。
3.2 C4位置的重要性前已述及, 用NH3+探针研究揭示了在C4附近有与正电荷相互作用的负电区, 推测若将C4的羟基置换成氨基, 可能提高与酶的亲和力, 因而合成了化合物3, 活性评价表明3显著提高了活性, Ki = 40 nmol·L-1, 是化合物2的100倍。
分子模拟研究了3与神经氨酸酶结合状态(图 3), 表明4位氨基被质子化, 带有正电荷, 可与Asp151和Glu119的羧基发生氢键结合, 距离分别为2.74 Å和2.96 Å (表明结合能很强)。此外, 水分子6X也与氨基发生氢键结合, 距离为2.77 Å。吡喃环上其他取代基的结合模式与1和2相同。

|
图 3 化合物3的4-氨基与神经氨酸酶形成氢键的示意图 |
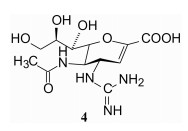
为了适配于GRID程序计算所揭示的C4有较大范围的正电荷结合区域, 将化合物3的4-氨基换成碱性更强、分布范围更大的胍基, 合成了化合物4, 对神经氨酸酶的活性比化合物3提高了40倍, Ki = 1 nmol·L-1, 提示基于酶和与底物结合的结构特征, 以及分子模拟获得的信息指导设计的有效性。由于碱性胍基(带有正电荷)结合的范围扩大, 可以同Glu119、Glu227、Asp151和Tyr277等多个氨基酸残基发生氢键和静电结合, 尤其重要的是胍基(正电荷)与Glu227的羧基(负电荷)发生电荷-电荷相互作用。此外, 还与水分子14X发生氢键结合。图 4是化合物4与神经氨酸酶分子对接所形成的氢键示意图。
|

|
图 4 化合物4与神经氨酸酶分子对接的示意图 |
化合物2~4的结构差异在于C4连接的基团不同, 比较3个化合物的C4基团与活性中心相关的氨基残基的结合的热力学性质, 计算出化合物4由于胍基的结合最强, 焓变最大, 化合物3的氨基结合次之, 焓变居中, 2的焓变最小, 3个化合物的焓变值与抑酶的活性值呈线性相关。表 1列出了化合物2~4的C4连接基与神经氨酸酶相关残基结合的焓变。
| 表 1 化合物2~4的C4基团与神经氨酸酶相关残基结合的焓变 |
化合物4体外对流感A和B病毒感染的细胞有显著抑制活性, 而对哺乳动物的神经氨酸酶不呈现抑制作用, 说明4具有选择性作用。因而确定为候选化合物, 定名扎那米韦(zanamivir)进入临床研究。由于口服生物利用度低, 给药途径是经鼻吸入, 这样气管和肺的吸收可达到15%。由于疗效的不确定性, FDA直到1999年才批准上市。然而, 这个由澳大利亚Monash大学和Biota公司研制、经葛兰素公司开发上市的首创性抗流感药物, 上市后并没有获得广泛的应用和认可, 数月后另一个上市的口服药物—神经氨酸酶抑制剂奥塞米韦(oseltamivir), 安全与有效性都优胜于扎那米韦。
4 后继的奥塞米韦在扎那米韦上市数月后, Gilead公司研制、罗氏公司开发的奥塞米韦经FDA批准上市, 后者虽不是首创, 但也是独立研发的。奥塞米韦可口服吸收、生物利用度高、代谢稳定等品质, 完胜于扎那米韦, 后来居上。
4.1 同一个起点奥塞米韦项目的启动, 也是基于神经氨酸酶的三维结构, 特别是从酶与唾液酸(1)和与Δ2唾液酸(2)的复合物三维结构出发, 实施了基于结构的药物设计(SBDD)。Gilead的研发目标是口服用药, 便于预防和治疗。
研究的切入点是确定母核, 用环己烯替换Δ2唾液酸(2)的二氢吡喃环, 环己烯的构象类似于过渡态的吡喃鎓离子的部分平面结构, 也与Δ2唾液酸的二氢吡喃构象相似。环己烯的优点在于:一是环的稳定性提高, 二是碳环上连接基团或侧链可以多样性和变换灵活性。
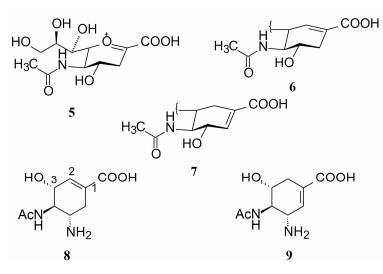
4.2 双键在环中的位置多取代环己烯的双键位置会影响活性强度, 因而具有特定性, 设计的位置对于模拟过渡态(5)是非常重要的。起初设计了双键在两个位置, 即类型6和7。6的双键位置类似于过渡态5, 7的双键位置类似于化合物2、3和4, 所以难于确定哪个双键位置最为适宜。为此合成了化合物8和9。
|
用流感神经氨酸酶评价8和9的活性, 8的活性IC50 = 6.3 μmol·L-1, 而9在200 μmol·L-1浓度下未呈现活性, 确定了环己烯的双键位置为Δ1, 2。
4.3 亲脂性侧链替换极性的甘油侧链分析化合物2与酶的晶体结构, 发现与C7相连的羟基并没有同酶发生结合作用, 因而可以去除这“限制的”7位羟基, C8和C9的羟基虽然同Glu276形成二齿型氢键结合, 但C8的CH与Arg224的亚烷基还发生疏水相互作用。这些分析促成了设计亲脂链的设计。化合物2或其类似物含有过多的极性基团, 这对于穿越细胞膜的吸收是不利因素, 所以将甘油侧链变为烷基链, 对于平衡分子的亲水-亲脂性应是有利的。此外, 唾液酸形成过渡态的氧鎓离子是缺电子状态, 在环己烯的3位(相当于吡喃环的6位)连接电负性强的氧原子以接近过渡态的缺电子性, 因而设计了C3以氧相连的烷基, 在化学合成上也容易实现。
4.4 环上的其他基团不变环己烯的其他位置如1位的羧基、4位的乙酰氨基、5位的氨基等基团与化合物3的配置相同, 设计依据与前述的是一样的。
4.5 C3-烷氧基的构效关系为了考察3-烷氧链的大小(长短)、形状(支化)和方向(构型)对活性的影响, 合成了化合物10~18, 活性评价的指标是对H1N1病毒的神经氨酸酶和病毒感染细胞生长的抑制活性, 分别用IC50和EC50表示。表 2列出了化合物的结构和活性。构效关系分析如下: ① R由H到正丙基(10~13)的抑制酶活性随碳原子增多而递增, 但延长成丁基(14)或异丁基(15)活性未见提高。② 甲基若连接在丙基的α位置(2-甲基丙基), 活性明显增加, 提高大约20倍。α位的支化映射了该部位有个疏水腔, 适配于α甲基的结合。α碳原子虽为手性碳, 但R (16)或S (17)构型的活性没有差异。③ α碳连接为乙基即支化呈对称的戊基(18)活性再提高10倍, 对感染的细胞也有高抑制活性。④ 继续增长碳链为庚氧基(19), 则活性降低, 提示该疏水腔不能容纳更大的亲脂性基团。⑤ 将化合物18的5-氨基变换为5-胍基, 仍保持高活性。比较化合物18与3和4 (扎那米韦)的活性, 提示18的活性显著高于化合物3, 与4 (扎那米韦)的活性相同(Kim CU, Lew W, Williams WA, et al. Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: design, synthesis, and structural analysis of carbocyclic sialic acid analogues with potent anti-influenza activity. J Am Chem Soc, 1997, 119: 681-690)。
| 表 2 化合物10~19的结构与抗流感神经氨酸酶和感染细胞的活性 |

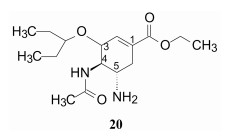
化合物18对A和B型流感神经氨酸酶具有高抑制活性, 而对哺乳动物的神经氨酸酶不显示抑制作用, 该选择性作用预示对人体的不良反应较少, 结合其他性质被确定为候选化合物。由于分子中含有羧基和脂肪氨基, 在生理条件下可形成内盐, 不利于细胞摄入和吸收。将18的羧酸乙酯化, 得到前药化合物20, 20本身没有活性(因为游离羧基对酶的结合至关重要), 却有利于吸收, 吸收后经肝脏酯酶水解, 释放出18而起效。化合物20命名为奥塞米韦(oseltamivir), 制成磷酸盐有利于溶解和吸收。
|
奥塞米韦的口服生物利用度F = 80%以上, 酯的血浆半衰期t1/2 = 1~3 h, 水解后释出的活性药物(游离酸18)的t1/2 = 6~10 h, 以18形式自尿中排出。奥塞米韦由罗氏公司开发, 经临床研究而确定了疗效, 与1999年经FDA批准上市, 成为预防和治疗流感的第一个口服药物(Davies BE. Pharmacokinetics of oseltamivir: an oral antiviral for the treatment and prophylaxis of influenza in diverse populations. J Antimicrob Chemother, 2010, 65: Suppl 2: ii5-10)。
4.7 奥塞米韦与神经氨酸酶的结合模式化合物18与神经氨酸酶复合物的晶体结构表明与预期的结合模式相同。如图 5所示, C1相连的羧基与3个精氨酸(Arg292、Arg371、Arg118) 的胍基形成强力结合。C5的氨基与Glu119和Asp151发生电荷-电荷样的氢键结合。C4上的乙酰氨基的甲基占据了由Trp178和Ile222构成的疏水腔, 酰基的氧原子与Arg152发生氢键结合。这些与前述的Δ2唾液酸(2)结合模式没有显著差别。不同的是C3侧链的结合模式。C3相连的3-戊氧基进入由Glu276、Ala246、Arg224和Ile222组成的疏水腔内, 这与扎那米韦的甘油片段的末端两个羟基与Glu276的羧基形成二齿样氢键结合不同。Glu276为了适配于疏水-疏水相互作用, 羧基的取向是向腔外, 亲脂性骨架参与了疏水结合。构效关系研究揭示的C3位烷基大小和形状对活性的影响, 佐证了3-戊基与疏水腔结合的熵效应达到最大值。

|
图 5 化合物18与神经氨酸酶的晶体结构 |