 2017, Vol. 52
2017, Vol. 52



2. 四川大学华西医学中心, 四川 成都 610041
2. West China Center of Medical Sciences, Sichuan University, Chengdu 610041, China
染色体重塑在DNA复制、修复, 以及调节表观遗传基因表达的过程中发挥着重要的作用[1]。DNA甲基化和组蛋白修饰是染色体重塑的主要表现形式。根据被修饰基团的不同, 组蛋白修饰包括乙酰化、甲基化和磷酸化。在转录前的组蛋白修饰中, 赖氨酸甲基化被研究的最为广泛[2]。组蛋白赖氨酸甲基化修饰是造成表观遗传变化的原因之一。
调节赖氨酸甲基化的酶主要分为两类:组蛋白赖氨酸甲基转移酶(histone lysine methyltransferases, HKMTs)和组蛋白赖氨酸去甲基化酶(histone lysine demethylases, HKDMs)。HKMTs负责将甲基从甲基供体转移到组蛋白赖氨酸上, 完成甲基化修饰。而HKDMs的功能主要是将甲基从组蛋白赖氨酸上移除, 完成脱甲基过程[3]。二者生物学功能相反, 共同调节着组蛋白赖氨酸的甲基化状态。
至今已发现的组蛋白赖氨酸去甲基化酶(HKDMs)[4]有两类:一类为赖氨酸特异性去甲基化酶(lysine specific demethylases, LSDs), 也称FAD (flavin adenine dinucleotide)依赖性胺氧化酶, 包括LSDl和LSD2。另一类为JMJD (Jumonji domain-containing protein)组蛋白去甲基化酶, 是一类分子中含有Jumonji结构域的蛋白家族, 包括KDM2/7、KDM3、KDM4、KDM5和KDM6, 其去甲基催化过程依赖于Fe(Ⅱ)和α-酮戊二酸(2-OG)的参与。
研究表明, 组蛋白赖氨酸去甲基化酶(HKDMs)的错误调控和诸多疾病的发生发展密切相关[5, 6], 如老年性疾病、肿瘤等。此外, 最新研究表明HKDMs还与肿瘤药物的耐药有关。因此, HKDMs已成为抗肿瘤新药开发的重要靶标, 越来越受到人们的关注[7, 8]。截止目前, 已有较多的HKDMs抑制剂被发现[4, 9, 10]。本文将对HKDMs的生物药理学作用以及其抑制剂进行综述, 并展望它们在疾病治疗方面的潜力。
1 LSDs的生物药理作用及其抑制剂 1.1 LSD1和LSD2的生物药理作用LSD1是哈佛医学院施扬课题组于2004年发现的第一个组蛋白赖氨酸特异性去甲基化酶1 (lysine specific demethylase 1, LSD1)[11]。该课题组首次确认组蛋白甲基化是一个动态平衡过程。这一发现对组蛋白修饰的作用机制及其相应的药物研究提供了全新思路。LSD1是一种黄素腺嘌呤二核苷酸依赖的去甲基化酶, 能够去除H3K4和H3K9的单、双甲基, 从而调节组蛋白和其他蛋白的相互作用, 并影响基因转录的激活、抑制和染色体失活等过程。
LSD1的底物, 除组蛋白H3外, 还有P53、DNA甲基转移酶1、STAT3[12]、E2F1和MYPT1[13]等, 通过去甲基化修饰, 调节相应细胞的生物学功能。研究报道, LSD1和许多转录因子结合, 调控基因的表达。表 1中总结了LSD1调控的靶基因和结合蛋白[4]。
| Table 1 Genes regulated by LSD1 |
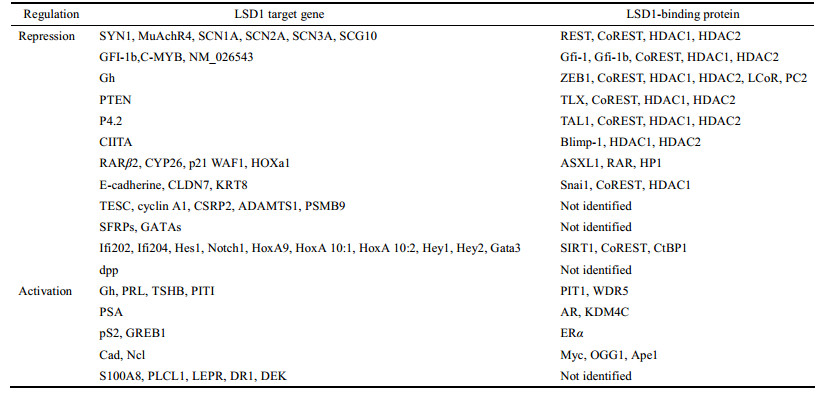
LSD1在多种癌症细胞和和癌组织中高表达[14, 15], 如成神经细胞瘤、成视网膜细胞瘤、前列腺癌、乳腺癌、肺癌和膀胱癌细胞。此外, RNAi介导的敲除实验以及LSD1抑制实验表明, LSD1酶和癌细胞的增殖有关, 主要是通过调控存活前基因的表达和P53基因转录活性。因此, LSDl已成为表观遗传学抗肿瘤药物的新靶点[16, 17], 高活性的LSD1抑制剂可作为潜在的抗癌药物, 用于相关癌症的治疗[18]。
此外, LSD1也能调控病毒基因的转录[19]。在单纯性疱疹病毒(HSV)和水痘-带状疱疹病毒(VZV)中, 病毒基因在宿主细胞中转录, 需要增加H3K4的甲基化水平, 同时降低H3K9甲基化水平。为了增加甲基化水平, 病毒需要募集宿主细胞因子1 (HCF-1) 和HKMT复合体, Liang等[19]研究发现LSD1能与HKMT复合体中的HCF-1相互作用, 进而开启H3K9的去甲基化, 最终降低H3K9的甲基化水平。因此, LSD1抑制剂能阻断LSD1的活性导致病毒基因转录过程被抑制, 有望开发为新的抗HSV和抗VZV的药物。
LSD2是另一个黄素依赖的赖氨酸去甲基化酶[20], 于2009年被人们发现, 关于它的研究目前并不多。有报道称, 在肿瘤产生和基因激活转录中, LSD2进行H3K4去甲基化有助于DNA甲基化[21]。也有报道称LSD2能抑制转录的发生[22], 同时抑制活性不依赖于它的去甲基化活性。
1.2 LSD1去甲基化的催化机制Yang等[23]测定了LSD1-CoREST-H3复合物的X-射线晶体结构, 阐明了组蛋白H3被识别结合的机制[24]。结构数据显示, 组蛋白H3有3个连续的γ-转角, 形成一个边链空间, 其N端被包入含有Asn、Trp和2个Asp残基的阴离子结合口袋。同时, 晶体结构也显示了被甲基修饰赖氨酸邻近FAD, 证实了LSD1去甲基化催化反应由FAD介导发生。
LSD1去甲基化催化反应机制(图 1)主要包含3个步骤:脱氢, FAD从甲基修饰的赖氨酸上脱掉一个氢, 生成亚胺阳离子和FADH2;加水, 在亚胺阳离子中加入一分子水; 脱CHO, 脱掉一分子CHO, 生成少一个甲基的赖氨酸。对于二甲基修饰的赖氨酸, 重复前面3个步骤, 去掉剩余的一个甲基。值得注意的是, LSD1仅能对单甲基、二甲基修饰的赖氨酸进行去甲基, 而不能对三甲基修饰的赖氨酸进行去甲基。LSD1去甲基化催化机制的提出为开发LSD1抑制剂奠定了基础。

|
Figure 1 Demethylation process of mono-or di-methylated lysine catalyzed by LSD1 |
LSD1是一个胺氧化酶, 它和单胺氧化酶(MAOs) A和B具有同源性(约17.6%等同性)[25]。因此, 单胺氧化酶的抑制剂也可以抑制LSD1。例如, 单胺氧化酶抑制剂化合物1对LSD1的抑制活性(Ki值)在1 mmol·L-1左右。化合物2 (反-2-苯基环丙烷胺, PCPA)和化合物3被报道对LSD1有抑制作用。但是它们对LSD1的抑制活性和选择性均不高(图 2), 因此怎样提高MAOs抑制剂对LSD1的活性和选择性值得进一步探讨研究。相信基于结构的药物设计将帮助找到答案。

|
Figure 2 Chemical structures of typical LSD1 inhibitors based on MAOs inhibitors |
化合物2被证明是一个LSD1的不可逆抑制剂。其抑制LSD1的机制主要是通过与FAD的黄素环发生加成反应, 形成共价加成物。尽管化合物2对LSD1的抑制活性较低, 但在高浓度下也能全面抑制LSD1的活性, 抑制成神经细胞瘤和膀胱癌细胞的增殖。研究证明, 化合物2不仅具有抗癌活性, 也具有抗病毒活性。通过对化合物2的结构改造, 衍生了一系列LSD1的抑制剂, 如化合物4~11 (图 3)[26, 27]。它们都表现出较好的LSD1抑制活性(Ki < 200 μmol·L-1), 尤其是化合物9, 其不仅具有最好的抑制活性(Ki = 0.61 μmol·L-1), 也具有对LSD1最好的选择性。在HEK293T细胞中, 化合物9浓度为1 μmol·L-1时, 明显观测到H3K4me2水平增高。通过观察这些化合物的特征, 发现在MAOs抑制剂(如化合物2)中增加一些大的疏水基团能增加对LSD1的选择性。

|
Figure 3 Chemical structures of compounds 4-11 |
化合物12、13是基于化合物1、3设计的具有小分子肽类结构的LSD1抑制剂, 通过引入小分子肽结构, 它们对LSD1的抑制活性明显提升[28]。同时也发现化合物13的活性明显大于化合物12 (13大约是12的25倍), 这与化合物1和3对LSD1的抑制活性关系表现一致, 说明极性较强的肼基(-NH-NH2)比疏水性较强的炔基具有更优的药效结构(图 4)。

|
Figure 4 Examples of small peptide-based LSD1 inhibitors |
基于多胺的LSD1抑制剂也被报道[29], 它们是基于LSD1和FAD依赖的多胺氧化酶的同源性设计的抑制剂, 如化合物14~16。化合物14是二双胍类化合物, 是LSD1的非竞争性的抑制剂(IC50约为1 μmol·L-1), 通过抑制LSD1的活性, 显著增加了H3K4me2水平, 最终抑制了直肠癌的增殖。化合物15是Sharma等[30]报道的硫脲类化合物, 它对LSD1抑制效果较好, 能明显增加H3K4的甲基化水平。在LSD1高表达的Calu-6细胞中, 化合物15对其增殖抑制活性(GI50)在9~40 μmol·L-1内。此外, 当化合物16和DNA甲基转移酶抑制剂联用处理人结肠癌肿瘤模型, 发现能显著抑制肿瘤的生长。化合物14~16的分子结构见图 5。

|
Figure 5 Examples of polyamine-based LSD1 inhibitors |
前面已提及, JMJD组蛋白去甲基化酶是另一类重要的组蛋白赖氨酸去甲基化酶[31], 包括KDM2/7、KDM3、KDM4、KDM5和KDM6, 它们参与许多基因的表达。例如, Klose等[32]发现KDM3A (另名: JHDM2A/JMJD1A)通过对H3K9me3和H3K36me3去甲基, 显著降低了ASH2 (achaete-scute complex homologue 2 gene)基因的表达。
研究表明, JMJD组蛋白去甲基化酶与许多恶性肿瘤的发生和耐药有关[33, 34], 如Kauffman等[35]发现KDM4A与膀胱癌的的发生和增生关系密切。KDM4C的基因敲除结果表明KDM4C与食管鳞癌、前列腺癌、乳腺癌、原发性纵隔B细胞淋巴瘤和霍奇金淋巴瘤的细胞增殖有关。因此, JMJD组蛋白去甲基化酶已经成为表观遗传的重要靶点, 受到越来越多的关注, 其抑制剂有望成为抗癌治疗的新型武器。
2.2 JMJD组蛋白去甲基化酶的催化机制JMJD组蛋白去甲基化酶的结构特征被广泛研究, 晶体结构的解析使得其结构和结合机制得以阐明[36], Jumonji结构域是其催化中心, 活性位点处α-酮戊二酸(2-OG)紧邻Fe(Ⅱ)。酶活性中心有3个保守的氨基酸, 分别是2个组氨酸(His)和1个谷氨酸(Glu), 催化过程依赖于O2的参与, 具体脱甲基催化机制见图 6。基于JMJD组蛋白去甲基化酶的催化原理和机制, 结合基于结构(structure-based)和基于底物(substrate-based)的药物设计方法, 药物研究者们发现了一系列的JMJD组蛋白去甲基化酶抑制剂。

|
Figure 6 Demethylation process of methylated lysine catalyzed by Jumonji domain-containing protein (JMJD) histone demethylases |
JMJD组蛋白去甲基化酶(简称JMJD去甲基化酶)抑制剂是对抗赖氨酸去甲基过程的重要物质, 是调节赖氨酸去甲基诱导的表观基因错误调控和基因异常表达的重要武器, 有望成为表观遗传疾病治疗的新型药物。目前, 根据分子结构和发现原理, JMJD去甲基化酶的抑制剂主要分为4类: α-酮戊二酸(2-OG)类似物、基于吡啶(或嘧啶)的JMJD去甲基化酶抑制剂、异羟肟酸类JMJD去甲基化酶抑制剂、多酚类JMJD去甲基化酶抑制剂及其他。
2.3.1 2-OG类似物2-OG是JMJD去甲基化酶脱甲基催化的重要底物, 在催化过程中扮演着重要的角色。通过分析JMJD去甲基化酶的晶体结构, 研究者发现了JMJD去甲基化酶活性中心氨基酸残基及2-OG与Fe(Ⅱ)的重要结合特征。随后, 基于底物的药物设计方法挖掘出许多2-OG类似物也具有较好的JMJD去甲基化酶抑制活性[37], 如化合物17~19 (图 7)。由于2-OG类似物是2-OG的竞争性抑制剂, 因此, 这类抑制剂普遍存在广谱、选择性差等特点。故该类抑制剂的设计不仅要考虑分子中与Fe(Ⅱ)的络合基团, 同时也要考虑酶活性位点处的空间特征, 增强其与活性位点临近氨基酸的作用力, 才能提高其酶抑制活性和选择性。

|
Figure 7 Examples of α-ketoglutarate (2-OG) analogue-based JMJD demethylases inhibitors. |
基于结构的药物设计(如分子对接)发现, 某些含羧基吡啶结构(或羧基嘧啶)的小分子化合物具有较好的JMJD去甲基化酶抑制活性, 如化合物20~25 (图 8)。这些抑制剂的结构中都含有氢键的受体(吡啶N或嘧啶N), 易于与酶活性中心的氢键供体形成氢键作用, 增加分子结合稳定性。其中, 化合物22是杨胜勇团队近期发现的具有羧基嘧啶结构的KDM5A的选择性抑制剂(IC50 = 0.22 μmol·L-1)[38], 是通过虚拟高通量筛选结合化学结构改造而得。该化合物在设计时充分考虑了其在酶活性口袋中的结合模式和空间位置, 最终表现出KDM5A的选择性。当用化合物22处理KDM5A高表达的人乳腺癌细胞株ZR-75-1后, 细胞中H3K4me3的含量明显增加, 且成浓度依赖关系。另外, 化合物25 (JIB-04) 是Wang等[39]通过细胞高通量筛选发现的JMJD去甲基化酶抑制剂, 它在体内体外都表现出良好的JMJD去甲基化酶的抑制活性, 并存在剂量依赖关系。在小鼠的H358肺癌细胞异种移植模型中, JIB-04能明显消除肿瘤生长。此外, 在4T1乳腺癌肿瘤模型中, JIB-04能显著增加其中位生存期。

|
Figure 8 Examples of pyridine or pyrimidine based JMJD demethylases inhibitors |
异羟肟酸具有双配位基(-N(OH)-C(O)-), 通过这种双配位基可以形成它的金属螯合物, 包括Fe(Ⅱ)。但单纯的螯合作用, 很容易导致小分子的脱靶效应产生, 故抑制剂在设计过程中还应充分考虑酶活性位点的空间结构和周围氨基酸残基的特征(带电性和疏水性)。基于这个原理, 异羟肟酸类JMJD去甲基化酶抑制剂被设计和发现, 例如化合物26~28 (图 9)。化合物26是基于KDM4A的晶体结构和KDM4C的同源模型设计而来, 其对KDM4A和KDM4C的IC50分别为0.8和2.2 μmol·L-1。化合物27是Luo等[40]基于结构的方法发现的羟肟酸类JMJD去甲基化酶抑制剂。研究发现, 在KDM4C高表达的食管癌KYSE150细胞株中, 化合物27的甲基化前体选择性地抑制JMJD去甲基化酶, 表现出对食管癌细胞增殖的抑制效果, 这表明异羟肟酸类JMJD去甲基化酶抑制剂在抗癌治疗上具有较大的潜力。化合物28 (伏立诺他, vorinostat)是组蛋白去乙酰化酶HDACs和DNA甲基转移酶抑制剂, 由于其分子中含异羟肟酸结构, 因此也具有JMJD去甲基化酶抑制活性。

|
Figure 9 Examples of typical hydroxamic acid based JMJD demethylases inhibitors |
多酚类化合物是指分子结构中有若干个酚性羟基的化合物, 包括在植物食物中发现的黄酮类、单宁类、酚酸类以及花色苷类化合物, 也包括化学合成的多羟基化合物, 如化合物29~32 (图 10)[41]。研究发现, 多酚类化合物具有JMJD去甲基化酶抑制活性, 但由于其极性大、广谱、体外细胞抑制活性低等特点, 其成药性低, 进一步开发难度大。例如, 化合物29 (俗名:红培酚)不仅具有JMJD去甲基化酶活性, 也能抑制甲基转移酶SETDB1, 同时具有较差的体外细胞抑制活性。因此, 怎样提升多酚类化合物的去甲基化酶活性和选择性, 一直以来都是药物化学家的难题。同时, 其较大的分子极性特征阻碍了细胞跨膜运输, 表现出较差的细胞活性。化合物32是Sekirnik等[42]发现的双硫仑类似物, 通过对KDM4A (又名: JMJD2A)活性位点中Zn离子的移除发挥抗KDM4A活性。这表明, 能拔除JMJD去甲基化酶活性位点中金属离子的化合物, 有望成为JMJD去甲基化酶的抑制剂。

|
Figure 10 Examples of typical polyphenols-based JMJD demethylases inhibitors |
HKDMs与许多疾病的发生和耐药有关, 是表观遗传学药物开发的重要靶点。目前, 已有较多的HKDMs抑制剂被相继发现, 但大都存在活性低、选择性差等缺点。因此, 迫切希望有更多高活性、高选择性的HKDMs酶抑制剂被发现, 这些抑制剂不仅可以作为小分子探针, 用于探索去甲基化酶的生物学功能, 也可作为新药治疗HKDMs相关的表观遗传疾病。随着晶体结构数据库的不断更新完善, HKDMs晶体结构日益增多, 新药研究的途径将更加广泛, 基于结构的药物设计将有助于发现更多的赖氨酸去甲基化酶抑制剂。研发高效、低毒、高选择性的HKDMs抑制剂是未来抗肿瘤治疗的新方向, 必将造福于人类健康。
| [1] | Blackledge NP, Klose RJ. Histone lysine methylation: an epigenetic modification?[J]. Epigenomics, 2010, 2: 151–161. DOI:10.2217/epi.09.42 |
| [2] | Li B, Carey M, Workman JL. The role of chromatin during transcription[J]. Cell, 2007, 128: 707–719. DOI:10.1016/j.cell.2007.01.015 |
| [3] | Trojer P, Reinberg D. Histone lysine demethylases and their impact on epigenetics[J]. Cell, 2006, 125: 213–217. DOI:10.1016/j.cell.2006.04.003 |
| [4] | Suzuki T, Miyata N. Lysine demethylases inhibitors[J]. J Med Chem, 2011, 54: 8236–8250. DOI:10.1021/jm201048w |
| [5] | Salminen A, Kaarniranta K, Kauppinen A. Hypoxia-inducible histone lysine demethylases: impact on the aging process and age-related diseases[J]. Aging Dis, 2016, 7: 180–200. DOI:10.14336/AD.2015.0929 |
| [6] | Pathak SS, Maitra S, Chakravarty S, et al. Histone lysine demethylases of JMJD2 or KDM4 family are important epigenetic regulators in reward circuitry in the etiopathology of depression[J]. Neuropsychopharmacology, 2017, 42: 854–863. DOI:10.1038/npp.2016.231 |
| [7] | Thinnes CC, England KS, Kawamura A, et al. Targeting histone lysine demethylases-progress, challenges, and the future[J]. Biochim Biophys Acta, 2014, 1839: 1416–1432. DOI:10.1016/j.bbagrm.2014.05.009 |
| [8] | Hojfeldt JW, Agger K, Helin K. Histone lysine demethylases as targets for anticancer therapy[J]. Nat Rev Drug Discov, 2013, 12: 917–930. DOI:10.1038/nrd4154 |
| [9] | Gale M, Yan Q. High-throughput screening to identify inhibitors of lysine demethylases[J]. Epigenomics, 2015, 7: 57–65. DOI:10.2217/epi.14.63 |
| [10] | Westaway SM, Preston AG, Barker MD, et al. Cell penetrant inhibitors of the KDM4 and KDM5 families of histone lysine demethylases. 2. pyrido[3, 4-d]pyrimidin-4(3H)-one derivatives[J]. J Med Chem, 2016, 59: 1370–1387. DOI:10.1021/acs.jmedchem.5b01538 |
| [11] | Shi Y, Lan F, Matson C, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1[J]. Cell, 2004, 119: 941–953. DOI:10.1016/j.cell.2004.12.012 |
| [12] | Yang J, Huang J, Dasgupta M, et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes[J]. Proc Natl Acad Sci U S A, 2010, 107: 21499–21504. DOI:10.1073/pnas.1016147107 |
| [13] | Cho HS, Suzuki T, Dohmae N, et al. Demethylation of RB regulator MYPT1 by histone demethylase LSD1 promotes cell cycle progression in cancer cells[J]. Cancer Res, 2011, 71: 655–660. DOI:10.1158/0008-5472.CAN-10-2446 |
| [14] | Wissmann M, Yin N, Müller JM, et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression[J]. Nat Cell Biol, 2007, 9: 347–353. DOI:10.1038/ncb1546 |
| [15] | Yokoyama A, Takezawa S, Schule R, et al. Transrepressive function of TLX requires the histone demethylase LSD1[J]. Mol Cell Biol, 2008, 28: 3995–4003. DOI:10.1128/MCB.02030-07 |
| [16] | Chen C, Wang Y, Wang S, et al. LSD1 sustains estrogen-driven endometrial carcinoma cell proliferation through the PI3K/AKT pathway via di-demethylating H3K9 of cyclin D1[J]. Int J Oncol, 2017, 50: 942–952. |
| [17] | Wang S, Zhao LJ, Zheng YC, et al. Design, synthesis and biological evaluation of[J]. Eur J Med Chem, 2017, 125: 940–951. DOI:10.1016/j.ejmech.2016.10.021 |
| [18] | Zheng YC, Yu B, Jiang GZ, et al. Irreversible LSD1 inhibitors: application of tranylcypromine and its derivatives in cancer treatment[J]. Curr Top Med Chem, 2016, 16: 2179–2188. DOI:10.2174/1568026616666160216154042 |
| [19] | Liang Y, Vogel JL, Narayanan A, et al. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency[J]. Nat Med, 2009, 15: 1312–1317. DOI:10.1038/nm.2051 |
| [20] | Karytinos A, Forneris F, Profumo A, et al. A novel mammalian flavin-dependent histone demethylase[J]. J Biol Chem, 2009, 284: 17775–17782. DOI:10.1074/jbc.M109.003087 |
| [21] | Katz TA, Vasilatos SN, Harrington E, et al. Inhibition of histone demethylase, LSD2(KDM1B), attenuates DNA methylation and increases sensitivity to DNMT inhibitor-induced apoptosis in breast cancer cells[J]. Breast Cancer Res Treat, 2014, 146: 99–108. DOI:10.1007/s10549-014-3012-9 |
| [22] | Yang Y, Yin X, Yang H, et al. Histone demethylase LSD2 acts as an E3 ubiquitin ligase and inhibits cancer cell growth through promoting proteasomal degradation of OGT[J]. Mol Cell, 2015, 58: 47–59. DOI:10.1016/j.molcel.2015.01.038 |
| [23] | Yang M, Culhane JC, Szewczuk LM, et al. Structural basis of histone demethylation by LSD1 revealed by suicide inactivation[J]. Nat Struct Mol Biol, 2007, 14: 535–539. DOI:10.1038/nsmb1255 |
| [24] | Forneris F, Binda C, Dall'Aglio A, et al. A highly specific mechanism of histone H3-K4 recognition by histone demethylase LSD1[J]. J Biol Chem, 2006, 281: 35289–35295. DOI:10.1074/jbc.M607411200 |
| [25] | Schmidt DM, McCafferty DG. trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1[J]. Biochemistry, 2007, 46: 4408–4416. DOI:10.1021/bi0618621 |
| [26] | Gooden DM, Schmidt DM, Pollock JA, et al. Facile synthesis of substituted trans-2-arylcyclopropylamine inhibitors of the human histone demethylase LSD1 and monoamine oxidases A and B[J]. Bioorg Med Chem Lett, 2008, 18: 3047–3051. DOI:10.1016/j.bmcl.2008.01.003 |
| [27] | Binda C, Valente S, Romanenghi M, et al. Biochemical, structural, and biological evaluation of tranylcypromine derivatives as inhibitors of histone demethylases LSD1 and LSD2[J]. J Am Chem Soc, 2010, 132: 6827–6833. DOI:10.1021/ja101557k |
| [28] | Szewczuk LM, Culhane JC, Yang M, et al. Mechanistic analysis of a suicide inactivator of histone demethylase LSD1[J]. Biochemistry, 2007, 46: 6892–6902. DOI:10.1021/bi700414b |
| [29] | Huang Y, Greene E, Murray Stewart T, et al. Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes[J]. Proc Natl Acad Sci U S A, 2007, 104: 8023–8028. DOI:10.1073/pnas.0700720104 |
| [30] | Sharma SK, Wu Y, Steinbergs N, et al. (Bis)urea and(bis)thiourea inhibitors of lysine-specific demethylase 1 as epigenetic modulators[J]. J Med Chem, 2010, 53: 5197–5212. DOI:10.1021/jm100217a |
| [31] | Suzuki T, Ozasa H, Itoh Y, et al. Identification of the KDM2/7 histone lysine demethylase subfamily inhibitor and its antiproliferative activity[J]. J Med Chem, 2013, 56: 7222–7231. DOI:10.1021/jm400624b |
| [32] | Klose RJ, Yamane K, Bae Y, et al. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36[J]. Nature, 2006, 442: 312–316. DOI:10.1038/nature04853 |
| [33] | Banelli B, Carra E, Barbieri F, et al. The histone demethylase KDM5A is a key factor for the resistance to temozolomide in glioblastoma[J]. Cell Cycle, 2015, 14: 3418–3429. DOI:10.1080/15384101.2015.1090063 |
| [34] | Hou J, Wu J, Dombkowski A, et al. Genomic amplification and a role in drug-resistance for the KDM5A histone demethylase in breast cancer[J]. Am J Transl Res, 2012, 4: 247–256. |
| [35] | Kauffman EC, Robinson BD, Downes MJ, et al. Role of androgen receptor and associated lysine-demethylase coregulators, LSD1 and JMJD2A, in localized and advanced human bladder cancer[J]. Mol Carcinog, 2011, 50: 931–944. DOI:10.1002/mc.20758 |
| [36] | Cloos PA, Christensen J, Agger K, et al. The putative oncogene GASC1 demethylates tri-and dimethylated lysine 9 on histone H3[J]. Nature, 2006, 442: 307–311. DOI:10.1038/nature04837 |
| [37] | Smith EH, Janknecht R, Maher LJ 3rd. Succinate inhibition of alpha-ketoglutarate-dependent enzymes in a yeast model of paraganglioma[J]. Hum Mol Genet, 2007, 16: 3136–3148. DOI:10.1093/hmg/ddm275 |
| [38] | Wu X, Fang Z, Yang B, et al. Discovery of KDM5A inhibitors: homology modeling, virtual screening and structure-activity relationship analysis[J]. Bioorg Med Chem Lett, 2016, 26: 2284–2288. DOI:10.1016/j.bmcl.2016.03.048 |
| [39] | Wang L, Chang J, Varghese D, et al. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth[J]. Nat Commun, 2013, 4: 2035. |
| [40] | Luo X, Liu Y, Kubicek S, et al. A selective inhibitor and probe of the cellular functions of Jumonji C domain-containing histone demethylases[J]. J Am Chem Soc, 2011, 133: 9451–9456. DOI:10.1021/ja201597b |
| [41] | Sakurai M, Rose NR, Schultz L, et al. A miniaturized screen for inhibitors of Jumonji histone demethylases[J]. Mol Biosyst, 2010, 6: 357–364. DOI:10.1039/B912993F |
| [42] | Sekirnik R, Rose NR, Thalhammer A, et al. Inhibition of the histone lysine demethylase JMJD2A by ejection of structural Zn(Ⅱ)[J]. Chem Commun(Camb), 2009: 6376–6378. |