 2017, Vol. 52
2017, Vol. 52



利奈唑胺(linezolid)化学名为N-[3-[3-氟-4-(吗啉-4-基)苯基]-2-氧代噁唑烷-5(S)-基甲基]乙酰胺, 化学结构见图 1, 是第一个应用于临床的新型噁唑烷酮类抗生素, 对革兰阳性菌以及多重耐药的病原体抗菌活性(包括耐药性结核杆菌)较好, 它通过阻断细菌蛋白质合成来完成抗菌作用, 因此不易与其他抗生素产生交叉耐药性[1, 2]。利奈唑胺主要用于治疗耐万古霉素粪肠球菌(VRE)引起的菌血症、耐甲氧西林金葡菌(MRSA)引起的院内肺炎和综合性皮肤感染以及青霉素敏感的肺炎链球菌(PRSP)引起的菌血症[3-5]。目前有片剂和注射液两种剂型。

|
Figure 1 Chemical structure of linezolid |
药物中的杂质种类及其含量与药物的安全性和有效性密切相关, 杂质的毒性是药品有关物质研究的关键内容之一[6]。文献报道HPLC法测定利奈唑胺原料药的有关物质[7, 8]和手性异构体[9], 对利奈唑胺片的杂质及其毒性的研究均未见报道。本文采用UFLC-MS/MS技术对原研企业辉瑞公司的利奈唑胺片的杂质进行了研究, 测定了利奈唑胺片的有关物质, 通过强制降解实验, 研究推断了其在强酸、强碱、氧化、水解等降解条件下的降解杂质结构, 对杂质的质谱裂解规律与途径进行了分析与归纳, 并采用计算机辅助化合物毒性预测系统对降解杂质进行了毒性预测。
材料与方法仪器 UFLC-20AD XR型超快速液相色谱仪(日本岛津公司), 配有二元泵、自动进样器和紫外检测器; AB Qtrap5500 (美国AB Sciex公司)质谱仪, 配有ESI电喷雾离子源、三重四级杆-线性离子阱质量分析器及Analyst1.6分析软件。XP205电子天平(瑞士Mettler公司); KQ2200DE型数控超声波清洗器(昆山市超声仪器有限公司); HPG-280B光照培养箱(哈尔滨市东联电子技术开发有限公司); DHG-9023A电热鼓风干燥箱(上海一恒科学仪器有限公司)。
毒性预测软件 有机化合物急性毒性在线预测系统(CISOC-WEBPAT)、有机化合物致癌毒性在线预测系统(CISOC-WEBPCT)和有机化合物致突变毒性在线预测系统(CISOC-WEBPMT)均由中国科学院上海有机化学研究所提供技术支持。
试药与试剂 利奈唑胺片(斯沃, 批号: L08931), 辉瑞公司; 利奈唑胺(linezolid), 批号HY-10394, 纯度 > 98%, 购自MCE公司。杂质1 (impurity 1), (S)-1-氨基-3-[(3-氟-4-吗啉基苯基)氨基]-2-丙醇, 批号L27415, 纯度98.29%, 购自SINCO公司; 杂质2 (impurity 2), (S)-5-(乙酰胺甲基)-3-(3-氟-4-氧代吗啉基苯基)噁唑烷酮, 批号L466530, 纯度99.43%, 购自TRC公司; 杂质3 (impurity 3), (S)-5-(氨甲基)-3-(3-氟-4-吗啉基苯基)噁唑烷酮, 批号F0L498, 纯度99.8%, 购自USP官网; 杂质4 (impurity 4), (R)-N-[3-[(3-氟-4-吗啉基苯基)氨基]-2-羟基丙基]乙酰胺, 批号1967-071A4, 纯度99.7%, 购自TLC公司。
乙腈色谱纯, 美国Fisher公司; 甲酸为色谱纯, CNW公司; 实验用水为超纯水, 其他试剂为分析纯。
对照品储备液 分别称取各杂质及利奈唑胺对照品适量, 精密称定, 加乙腈制成质量浓度分别为0.5 mg·mL-1的溶液, 摇匀备用。
样品溶液 精密称取利奈唑胺片粉末适量(相当于利奈唑胺50 mg)于100 mL量瓶中, 精密加乙腈适量, 超声溶解, 用乙腈稀释至刻度, 滤过, 取续滤液, 即得。
色谱条件 色谱柱: Shim-pack XR-ODS (75 mm × 3 mm, 2.2 μm); 柱温: 30 ℃, 流动相: A相为0.1%甲酸水溶液, B相为0.1%甲酸乙腈溶液; 梯度洗脱(0~3 min, 95% A; 3~10 min, 95%~65% A; 10~15 min, 65%~50% A; 15~17 min, 50%~10% A; 17~17.3 min, 10%~95% A), 流速: 0.4 mL·min-1, 进样量: 3 μL, 测定波长: 254 nm。
质谱条件 离子源:电喷雾离子化(ESI); 检测方式:正离子模式; CUR: 20 psi (1 psi ≈ 6.9 kPa); CAD: High; IS: 5.5 kV; TEM: 450 ℃; GS1: 60 psi; GS2: 40 psi; EP: 10 V; CXP: 10。质量扫描范围: 50~500 Da; 扫描速度: 10 000 Da·s-1; 实时调节每个化合物的DP和CE。
强制降解 精密称取利奈唑胺片粉末进行强制降解实验。① 酸降解实验:称取适量(相当于利奈唑胺5 mg)置10 mL量瓶中, 加入1 mol·L-1 HCl溶液1 mL, 80 ℃水浴10 min, 取出放至室温, 用1 mol·L-1 NaOH中和后加乙腈稀释至刻度, 摇匀。② 碱降解实验:称取适量(相当于利奈唑胺5 mg)置10 mL量瓶中, 加入1 mol·L-1 NaOH溶液1 mL, 80 ℃水浴10 min, 取出放至室温, 用1 mol·L-1 HCl中和后加乙腈溶液稀释至刻度, 摇匀。③ 氧化降解实验:称取适量(相当于利奈唑胺5 mg)置10 mL量瓶中, 加入30% H2O2溶液1 mL, 放置8 h, 振摇, 加乙腈稀释至刻度。④ 加热水解:称取适量(相当于利奈唑胺5 mg)置10 mL量瓶中, 加入水1 mL, 80 ℃水浴8 h, 取出放至室温, 加乙腈稀释至刻度。⑤ 光照:取利奈唑胺片粉末于5000 lx光照24 h后, 称取适量(相当于利奈唑胺5 mg), 置10 mL量瓶中, 加乙腈溶解并稀释至刻度, 摇匀。⑥ 高温:取利奈唑胺片粉末在130 ℃放置24 h后, 称取适量(相当于利奈唑胺5 mg), 置10 mL量瓶中, 加乙腈溶解并稀释至刻度, 摇匀。所有强制降解实验溶液均用0.22 mm滤膜过滤, 取续滤液进行测定。同时随行各强制降解实验的空白。
线性关系 精密量取各杂质和利奈唑胺对照品的储备液1 mL置于10 mL量瓶中, 加乙腈稀释至刻度, 摇匀, 作为混合对照品储备液。取混合对照品储备液适量, 用乙腈定量稀释, 制成不同浓度的系列混合对照品溶液, 按色谱条件进样测定, 记录色谱图。
精密度 取各杂质对照品溶液适量, 加乙腈稀释制成浓度分别为5 μg·mL-1的混合杂质溶液, 按色谱条件重复测定6次, 计算各杂质峰面积的RSD (%)。
杂质的毒性预测 利用计算机辅助化合物毒性预测系统对降解杂质进行了急性毒性(生物测试模型是“rat, oral”)、致癌毒性(生物测试模型是“rat, oral”)和致畸变毒性(生物测试模型是“Salmonella typhimurium”)的预测。记录预测结果。
结果与讨论 1 UFLC测定杂质 1.1 样品溶液测定取样品溶液按UFLC色谱条件测定, 样品溶液未检出杂质(图 2)。

|
Figure 2 Chromatograms of sample solution and degradation solutions. a: Linezolid; b: Hydrolysis destruction; c: Acid destruction; d: Oxidation destruction; e: Base destruction. Peaks 1-4: Impurity 1-4 |
取强制降解实验溶液按UFLC色谱条件测定, 共产生4个杂质, 各降解杂质能与主峰良好分离(图 2)。按保留时间先后顺序将其编号为杂质1~杂质4。利奈唑胺片在固体状态下, 对高温、光照稳定; 在强酸、强碱、氧化及水溶液加热条件下, 均有不同程度的降解; 在碱降解条件下产生的两个杂质, 保留时间分别为2.1 min (1.1%, 杂质1)、7.0 min (87.8%, 杂质4), 在氧化降解条件下产生一个杂质, 保留时间为5.4 min (3.7%, 杂质2), 在酸降解条件下产生的两个杂质, 保留时间分别为6.3 min (1.6%, 杂质3)、7.0 min (0.54%, 杂质4), 加热水解产生一个杂质, 保留时间为7.0 min (0.25%, 杂质4)。
2 降解杂质结构分析采用建立的UFLC-MS/MS法对利奈唑胺降解溶液进行分析, 得到杂质1~杂质4的质谱图(图 3)。

|
Figure 3 The MS/MS spectra of the degradation impurities. A-D: Impurity 1-4 |
杂质1为碱降解条件下产生的杂质, 保留时间为2.1 min。在一级质谱中的准分子离子[M+H]+为m/z 270, 比利奈唑胺分子量小68 Da; 二级质谱碎片离子主要有m/z 235、209、196和163。质量数为m/z 235的碎片是准分子离子丢失中性分子H2O和NH3后生成的; 准分子离子分别丢失-C2H7NO和-C3H8NO·后得到质量数为m/z 209和196的碎片离子; 准分子离子也会先失去一个吗啉环, 接着再丢失HF中性分子得到质量数为m/z 163的碎片离子。杂质1为利奈唑胺在碱性条件下噁唑烷酮环和侧链上甲酰胺基同时断开的产物, 其一级质谱和二级质谱碎片与(S)-1-氨基-3-[(3-氟-4-吗啉基苯基)氨基]-2-丙醇相同, 且紫外光谱图也一致。杂质1的质谱裂解途径见图 4。

|
Figure 4 The plausible fragmentation pathway of impurity 1 |
杂质2为利奈唑胺在氧化降解条件下产生的杂质, 保留时间为5.4 min。其在一级质谱中的准分子离子[M+H]+为m/z 354, 比利奈唑胺分子量大16 Da; 二级质谱碎片离子主要有m/z 337、312、293、234、209和196。准分子离子先分别丢失-OH·和-C2H2O得到质量数为m/z 337和m/z 312的碎片, 接着m/z 337丢失CO2中性分子得到质量数为m/z 293的碎片。质量数为m/z 234的碎片离子来源于m/z 293, 因为丢失-C2H5NO片段, 质量数为m/z 209和m/z 196的碎片离子来源于m/z 312, 因为分别丢失(-O-C3H5NO2)和(-O-C4H6NO2·)片段。杂质2是利奈唑胺加了一个O原子的氧化产物, 由利奈唑胺的结构特点可知, 其最容易的加氧位点应该是吗啉环上的N原子。推测杂质2是利奈唑胺氮氧化物, 其与(S)-5-(乙酰胺甲基)-3-(3-氟-4-氧代吗啉基苯基)噁唑烷酮的一级图谱和二级质谱碎片相同, 且紫外光谱图一致。杂质2的质谱裂解途径见图 5。

|
Figure 5 The plausible fragmentation pathway of impurity 2 |
杂质3为利奈唑胺在酸降解条件下产生的杂质, 保留时间为6.27 min。其在一级质谱中的准分子离子[M+H]+为m/z 296, 比利奈唑胺分子量小42 Da; 二级质谱碎片离子主要有m/z 252、241、235、223、209、196和181。准分子离子丢失一个CO2中性分子生成质量数为m/z 252的碎片离子, 接着分别丢失NH3中性分子和-C3H7N2·得到质量数为m/z 235和m/z 181的碎片; 准分子离子峰也分别丢失-C3H5NO2和-C4H6NO2·得到质量数为m/z 209和m/z 196的碎片。质量数为m/z 241的碎片离子来源于准分子离子m/z 296, 因为连续丢失CO和HCN中性分子, 接着m/z 241又丢失一个H2O分子得到质量数为m/z 223的碎片离子。杂质3是利奈唑胺侧链上的甲酰胺基断裂的产物, 其与(S)-5-(氨甲基)-3-(3-氟-4-吗啉基苯基)噁唑烷酮的一级图谱和二级质谱碎片相同, 且紫外光谱图一致。杂质3的质谱裂解途径见图 6。

|
Figure 6 The plausible fragmentation pathway of impurity 3 |
杂质4为利奈唑胺在酸、碱、水解三个降解条件下均产生的杂质, 保留时间为7.0 min。在一级质谱中的准分子离子[M+H]+为m/z 312, 比利奈唑胺分子量小26 Da; 二级质谱碎片离子主要有m/z 295、235、215、209、196、191和116。准分子离子丢失-OH·生成质量数为m/z 295的碎片, 也分别丢失-C4H9NO2和C5H10NO2·得到质量数为m/z 209和m/z 196的碎片; 碎片m/z 116是准分子离子丢失196 Da碎片得到的; 准分子离子连续失去-C2H5NO和一个H2O中性分子得到质量数为m/z 235的碎片, 接着再分别丢失HF和-C2H4O得到质量数分别为m/z 215和m/z 191的离子碎片; 杂质4是利奈唑胺噁唑烷酮开环的产物, 其与(R)-N-[3-[(3-氟-4-吗啉基苯基)氨基]-2-羟基丙基]乙酰胺的一级图谱和二级质谱碎片相同, 且紫外光谱图一致。杂质4的质谱裂解途径见图 7。

|
Figure 7 The plausible fragmentation pathway of impurity 4 |
以利奈唑胺及各杂质质量浓度C为横坐标, 峰面积A为纵坐标, 绘制标准曲线, 得回归方程; 以利奈唑胺与各杂质的标准曲线斜率之比计算校正因子(表 1)。结果显示:杂质1和杂质2的校正因子分别为1.3和1.4;杂质3和杂质4的校正因子分别为0.9和1.1。
| Table 1 The regression equation and calibration factor of linezolid and impurities |
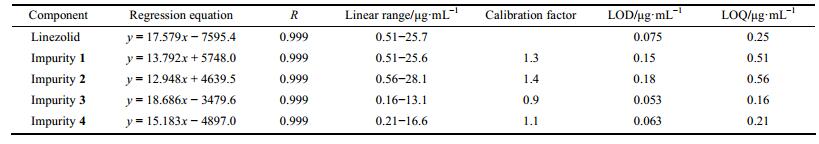
以信噪比3为检出限, 信噪比10为定量限。结果各杂质的检出限(LOD)和定量限(LOQ)如表 1所示。
3.3 精密度杂质1、杂质2、杂质3和杂质4峰面积的RSD (n = 6) 依次为1.2%、1.1%、1.2%和1.2%。
根据各杂质校正因子的测定结果, 杂质3和杂质4的校正因子在0.9~1.1范围内, 其测定可采用杂质对照品法或不加校正因子的主成分自身对照法计算; 杂质1和杂质2的校正因子不在0.9~1.1内, 应采用杂质对照品法或加校正因子的主成分自身对照法计算; 其他杂质采用不加校正因子自身对照法计算。
4 杂质的毒性预测结果表明: 4个降解杂质的急性毒性剂量LD50均为500~4 999 mg·kg-1 (rat, oral); 杂质1、杂质2和杂质4的致癌毒性预测结果为阳性, 其不致癌的可能性较低, 杂质3的预测结果为临界值; 杂质1和杂质2的致突变毒性预测结果为阳性, 其可能致突变, 杂质3和杂质4的致突变预测结果为阴性。
结论本文采用UFLC-MS/MS技术对利奈唑胺片剂的杂质进行了研究, 测定了利奈唑胺片剂的有关物质, 推断了其在酸、碱、氧化、水解等降解条件下的杂质结构, 并对杂质的质谱裂解规律与途径进行了分析与归纳。由降解实验可知, 利奈唑胺对高温、光照比较稳定, 在强酸、强碱、氧化、加热水解条件下, 均有不同程度的降解; 对杂质的降解机制分析, 利奈唑胺的降解发生在3个位点:噁唑烷酮开环水解、侧链甲酰基断裂、吗啉环上的N原子发生氧化。由降解产物的校正因子可知, 杂质1和杂质2与主成分的响应值相差较大, 如采用主成分自身对照法计算, 不能准确测定其含量; 对杂质1和杂质2应采用杂质对照品法或加校正因子的主成分自身对照法测定。
由降解杂质的毒性预测结果可知, 4个降解杂质均有可能致癌, 且杂质1和杂质2的致突变可能性也较高, 提示降解杂质的毒性需进一步的研究, 并加以控制。药品在生产加工、运输贮存过程中, 应尽量避免强酸、强碱、氧化和高温高湿因素的影响, 避免降解杂质的产生, 以保证药品的使用安全。
致谢: 中国科学院上海有机化学研究所(计算机化学和化学信息学研究室)姚建华教授对杂质毒性预测给予技术支持。| [1] | Clemtt D, Markham A. Linezolid[J]. Drugs Fut, 2000, 59: 815–827. |
| [2] | Utpal P, Yong PY, Franks WH, et al. Oxazolidinones mechanism of action: inhibition of the first peptide bong formation[J]. J Biol Chem, 2001, 276: 37199–37205. DOI:10.1074/jbc.M102966200 |
| [3] | Ament PW, Jamshed N, Horne JP. Linezolid: its role in the treatment of Gram-positive, drug-resistant bacterial infections[J]. Am Fam Physician, 2002, 65: 663–671. |
| [4] | Li Z, Willke RJ, Pintol A, et al. Comparison of length of hospital stay for patients with known or suspected methicillin-resistant Staphylococcus species infections treated with line zolid or vancomycin: a randomized, multicenter trial[J]. Pharmacotherapy, 2001, 21: 263–274. DOI:10.1592/phco.21.3.263.34198 |
| [5] | Locke JB, Finn J, Hilgers M, et al. Structure-activity relationships of diverse oxazolidinones for linezolid-resistant Staphylococcus aureus strains possessing the cfr methyltrans ferase gene or ribosomal mutations[J]. Antimicrob Agents Chemother, 2010, 54: 5337–5343. DOI:10.1128/AAC.00663-10 |
| [6] | Pan C, Liu F, Motto M. Identification of pharmaceutical impurities in formulated dosage forms[J]. J Pharm Sci, 2011, 100: 1228–1259. DOI:10.1002/jps.22376 |
| [7] | Lin XB. Determination of related substance in linezolid by HPLC[J]. Sci Time (科学时代), 2015, 8: 41–42. |
| [8] | Cao HY, Zhu YJ, Sang ZP, et al. Structure identification of the main degradation products of linezolid[J]. Chin J Pharm Anal (药物分析杂志), 2013, 33: 1342–1358. |
| [9] | Raju TS, Kutty OV, Ganesh V, et al. A validated stability-indicating LC method for the separation of enantiomer and potential impurities of linezolid using polar organic mode[J]. J Pharm Anal, 2012, 2: 272–278. DOI:10.1016/j.jpha.2012.03.006 |