 2016, Vol. 52
2016, Vol. 52



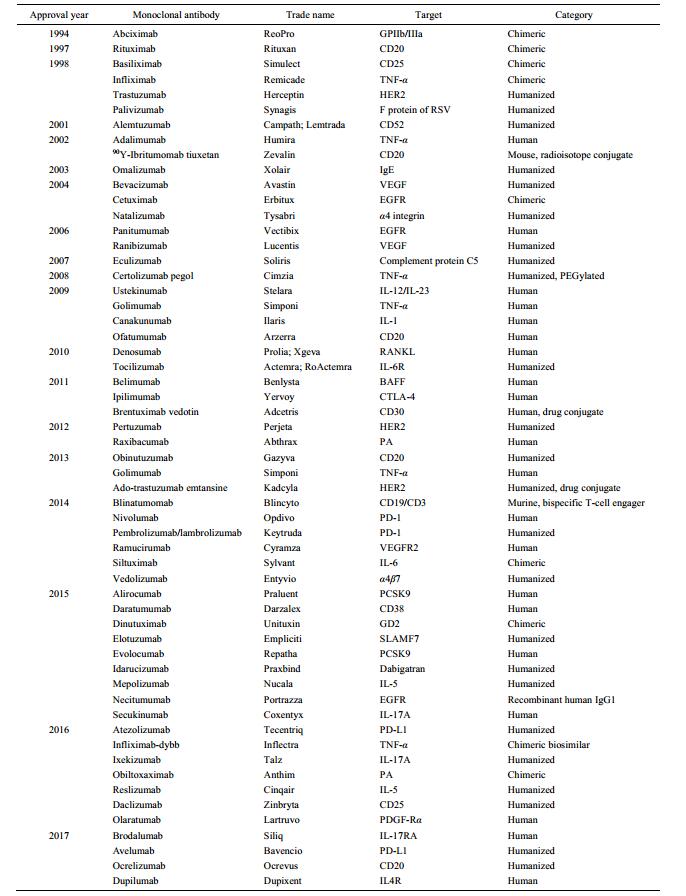
抗体是一种由效应B细胞分泌的免疫球蛋白, 协助免疫系统鉴别、中和抗原或其他外来物质。抗体由对称的2条重链及2条轻链组成, 其中重链类型决定抗体亚型, 抗原结合结构域(Fab)是重链和轻链的可变区组合形成的抗原结合位点[1]。迄今为止, 美国FDA已经获准50多种单克隆抗体上市用于肿瘤、自身免疫病等多种疾病的治疗(表 1)[2-4]。抗体的研究技术进展带动了靶向肿瘤和免疫分子单克隆抗体的研发,本文重点阐述单克隆抗体在临床疾病的诊断和治疗中的重要作用(图 1)。
| Table 1 Approved monoclonal antibody by FDA in market |

|
Figure 1 Classification and mechanism of therapeutic monoclonal antibodies (mAbs). A: mAbs recognize tumor specific antigen, bind to target tumor cells, then result in the direct cancer cells killing, or mediate antibody-dependent cellular cytotoxicity (ADCC) by immune cells, or induce complement-directed cytotoxicity (CDC). mAbs can also inhibit tumor progression by blocking cytokines, chemokines, toxic antigens (B) or immune checkpoint signaling pathways (C). Radioimmunoconjugates (D) and antibody-drug conjugates (E) deliver radioisotopes or potent toxic drugs to cancer cells, respectively. F: Bispecific antibodies bind activating antigens on immune cells with one arm and cancer cells with the other arm, and facilitate immune effector cells towards cancer cells |
自然界中有机体通过多克隆免疫应答对抗抗原。抗原由多个抗原决定簇组成, 这些抗原决定簇刺激机体产生多样混合的单克隆抗体即为多克隆抗体。此外, 同一种抗原决定簇也可刺激机体产生IgG、IgM、IgA、IgE和IgD等5类抗体。
1975年, Kohler和Milstein首先建立了杂交瘤技术。这种技术通过体外持续培养骨髓瘤细胞, 融合可分泌抗体的B淋巴细胞, 再经次黄嘌呤氨基蝶呤胸苷筛选及克隆化得到大量单克隆细胞株。这一技术为治疗性单克隆抗体的发展奠定基础[5]。根据单克隆抗体来源可分为鼠源化单克隆抗体(-omab, -莫单抗)、人鼠嵌合体单克隆抗体(-ximab, -昔单抗)、人源化单克隆抗体(-zumab, -组单抗)和全人源化单克隆抗体(-umab, -人单抗)。80年代末期, 鼠源单克隆抗体开始进入临床。然而, 在应用的过程中发现鼠源抗体存在明显不足, 包括过敏反应、诱导形成抗抗体、半衰期短及与人Fc受体弱结合[6]。更为重要的是, 鼠源抗体的直接作用、抗体依赖性细胞毒性(antibody-dependent cell-mediated cytotoxicity, ADCC)和补体依赖性细胞毒性(complement dependent cytotoxicity, CDC)相对较弱, 限制临床尤其是肿瘤治疗的应用[7]。为克服固有的免疫原性, 同时增强单克隆抗体效用, 基因工程技术发展出新的嵌合鼠-人单克隆抗体。通过基因工程手段可将鼠单克隆抗体的抗原特异性可变区移植到人单克隆抗体的恒定区, 此手段可使鼠单克隆抗体分子达到约65%的人源化[8]。嵌合单克隆抗体不仅免疫原性较低, 同时在人体内具有更长的半衰期[9]。为了更大限度地提高单克隆抗体效用, 通过移植鼠源抗体的高变区到人源抗体框架构建的人源化单克隆抗体同源性可达95%[10], 可以克服鼠源抗体和嵌合抗体的免疫原性。人源化单克隆抗体主要局限是其改造过程复杂耗时[1]。体外噬菌体展示技术和携带人可变区的转基因动物出现使得全人源单克隆抗体成为可能。人源化抗体和全人源抗体不仅免疫原性明显降低, 同时表现出与人源IgGs类似性质[11]。噬菌体展示技术在先导化合物优化过程中能够分离控制单克隆抗体的特异性和亲和力, 结合计算模拟等抗体亲和力成熟技术, 噬菌体展示技术在单克隆抗体的研发过程中发挥越来越重要的作用。
2 靶向肿瘤相关分子单克隆抗体进展迄今为止, 治愈肿瘤仍然是临床治疗面临的挑战。当手术、放疗和化疗无法控制肿瘤的发展进程, 抗体成为肿瘤治疗的重要辅助手段。与传统药物相比, 治疗性抗体不仅可以反复给药, 同时具有高靶向性、低毒副作用和疗效好等特征。本文根据单克隆抗体作用特点, 通过抑制肿瘤生存的关键分子、激活针对肿瘤的免疫固有性和适应性及抗体偶联细胞毒药物3个方面介绍单克隆抗体在肿瘤治疗领域的应用[12]。
2.1 抑制肿瘤生存的关键分子 2.1.1 CD20B淋巴细胞表面标志物CD20初始表达于Pro-B细胞(CD45R+, CD117+), 随着B细胞的成熟表达不断增加。CD20在分化成熟的浆细胞、正常造血干细胞及其他类型造血细胞系均无表达。因此, CD20成为治疗B淋巴细胞白血病的分子药靶。1997年, FDA批准罗氏集团开发的利妥昔单抗(rituximab, 美罗华)用于临床肿瘤治疗[13]。利妥昔单抗是一种靶向CD20的嵌合型单克隆抗体, 不仅能够直接抑制B细胞增殖、诱导CD20+ B细胞凋亡, 同时可通过ADCC和CDC杀死肿瘤细胞[14]。
2002年, 替伊莫单抗(90Y-ibritumomab tiuxetan, Zevalin)上市[15]。钇(90Y)标记抗CD20替伊莫单抗是一种典型放射性同位素偶联物。替伊莫单抗除促进B细胞凋亡, 亦可募集放射性核素如α粒子或β粒子进入药靶, 进一步增强肿瘤细胞杀伤效果。2003年, FDA批准Bexxar公司研发的放射-免疫偶联托西莫单抗(131I-tositumomab)。由于131I具有廉价、易获取和易与单抗结合等特点, 托西莫单抗在临床的应用更为广泛。2009年全人源化ofatumumab (HuMax-CD20, Arzerra)上市用于治疗慢性淋巴细胞白血病(CLL)。Ofatumumab的CD20结合表位与利妥昔单抗完全不同, 亲和力更强, 亦是通过ADCC和CDC途径发挥作用。
2013年, 罗氏公司研发的首个Ⅱ型糖基化的二代全人源抗CD20单克隆抗体obinutuzumab (afutuzumab, GA101, Gazyva)上市[16]。Obinutuzumab单抗属于无岩藻糖基抗体。在抗体制备的过程中过表达N-乙酰氨基葡萄糖转移酶3和高尔基体α-甘露糖苷酶Ⅱ (Golgi mannosidase 2), 降低抗体海藻糖含量, 进而增强ADCC效应[17, 18]。Obinutuzumab与CD20的结合表位与利妥昔单抗存在重叠区域[19], 也具有直接诱导细胞死亡的作用。研究发现, obinutuzumab对CLL和非霍奇金淋巴瘤(NHL)细胞的抑制活性强于利妥昔单抗。2017年美国罗氏基因泰克公司人源化抗体(90%~95%人源化) ocrelizumab上市用于治疗多发性硬化症。Ocrelizumab比第一代抗CD20单抗rituxan治疗效果明显, ADCC作用强, 免疫原性低。用药时间间隔长的特点显著改善患者依从性。
2.1.2 HER2HER2是一种具有酪氨酸激酶活性的跨膜糖蛋白, 属于表皮生长因子受体(HER)家族成员之一。HER家族包括HER1 (erbB1, EGFR)、HER2 (erbB2, NEU)、HER3 (erbB3) 及HER4 (erbB4)。HER家族成员在细胞生理过程中发挥重要调节作用, 通常经配体结合或相互之间形成二聚体介导信号转导[20]。研究发现, HER2不仅与肿瘤的发生发展密切相关, 同时也可作为重要的乳腺癌预后判定指标[13, 21]。靶向HER2的单克隆抗体能够下调HER2表达水平并抑制肿瘤生长。1998年, 美国基因泰克公司研发的人源化抗HER2抗体曲妥珠单抗(trastuzumab, Herceptin)结合HER2直接阻断表皮生长因子—HER2信号传导通路, 抑制肿瘤生长。2012年, FDA批准罗氏制药研发的人源化抗HER2抗体帕妥珠单抗(pertuzumab, Perjeta)用于治疗HER2阳性的转移性乳腺癌。帕妥珠单抗结合HER2后阻断HER2受体与其他HER家族受体二聚化, 进而抑制肿瘤生长和转移。
2.1.3 VEGF/VEGFR2血管内皮生长因子(VEGF)在诱导血管发生和生成、增强血管渗透性、内皮细胞生长、促进细胞迁移及抑制细胞凋亡等方面发挥关键作用。因此, VEGF及其受体VEGFR2成为肿瘤治疗的药物靶点[22]。2004年, FDA获准基因泰克公司研发的靶向VEGF贝伐珠单抗(bevacizumab, Avastin)治疗转移性结肠癌。重组人源贝伐珠单抗IgG1包含结合VEGF鼠源单抗的互补决定区以及人源抗体框架结构区。贝伐珠单抗此后扩展应用于肺癌、乳腺癌、肾癌及老年性黄斑变性等眼部疾病。2014年, Cyramza公司研发的抗VEGFR2单克隆抗体雷莫芦单抗(ramucirumab)上市。雷莫芦单抗阻断配体VEGF-A、VEGF-C和VEGF-D与受体VEGFR2的结合, 可有效阻断肿瘤的血供, 进而达到抑制肿瘤生长的目的。临床主要用于治疗目前最常见且致命的晚期转移癌, 包括胃癌、非小细胞型肺癌及结直肠癌。
2.1.4 EGFREGFR (HER1, cErbB-1) 属于HER家族。EGFR不仅调节人体正常细胞生长, 肿瘤发生后也促进肿瘤细胞增殖。表皮生长因子(EGF)和转化生长因子α (TGF-α)活化膜受体EGFR酪氨酸激酶, 促进EGFR构象变化形成二聚体, 激活下游信号包括Ras、Raf和PI3K-AKT等[23]。临床研究发现, 通过抗体阻断EGFR可以抑制直肠癌等肿瘤细胞通过上述信号通路引发的血管再生、肿瘤转移及耐药[23]。2004年, 德国默克雪兰诺公司研发的嵌合型IgG1西妥昔单抗(cetuximab, Erbitux)用于治疗转移性结直肠癌、转移性非小细胞肺癌和头颈部癌症。西妥昔单抗不仅可以阻断生长因子与EGFR相互作用, 同时也直接阻断突变EGFR配体非依赖的信号通路, 进而获得抑制肿瘤发生发展的效用。2006年, FDA批准美国安进(Amgen)公司研发的全人源化帕尼单抗(panitumumab, Vectibix)用于EGFR阳性转移性结肠癌。尽管IgG2型帕尼单抗比IgG1型西妥昔单抗人源化程度高、更易结合EGFR并阻断EGF或TGFα配体与EGFR结合, 抑制其酪氨酸激酶活性及肿瘤细胞增殖, 但是在转移性结肠癌的临床研究中并没有发现显著差别。
2.1.5 CD19CD19、CD21和CD81均为B淋巴细胞特异表面标志, 常被用作B淋巴细胞白血病的诊断标志。研究发现, 淋巴细胞根据不同抗原表达高低进行增殖和分化。低亲和力抗原受体协助B细胞对各种抗原保持特异性和敏感性。CD19从最早期可识别的B谱系细胞到B细胞母细胞的过程中维持表达, 直至成熟浆细胞时消失[24]。2014年, Amgen公司研发的博纳吐单抗(blinatumomab, Blincyto)上市用于治疗费城染色体阴性复发性急性淋巴细胞白血病。Blinatumomab是一种针对CD19/CD3双靶点的鼠源双特异性抗体, 通过B细胞的CD19识别位点和T细胞的CD3识别位点, 在B淋巴肿瘤细胞周围大量募集肿瘤患者自身T细胞, 激活T细胞发挥细胞毒作用[25]。
2.1.6 CD25CD25是由IL2RA基因编码的IL-2受体α链。高亲和力IL-2受体由α链(IL2RA)、β链(IL2RB)以及γ链(IL2RG)组成; β链同源二聚体形成中等亲和力IL-2受体; α链同源二聚体形成低等亲和力IL-2受体。CD25通常表达于活化T、B细胞、部分胸腺细胞和髓系前体细胞等。CD25已成为小鼠CD4+ FoxP3+ Treg细胞表面标志物。但是, 人体内发现大部分静息记忆T细胞组成性表达CD25[26]。CD25在大多数B淋巴细胞瘤、部分急性非淋巴细胞白血病、神经母细胞瘤、肥大细胞增多症以及肿瘤浸润性淋巴细胞中表达, 能够作为Ⅰ型人类T淋巴细胞白血病病毒受体发挥作用。1997年, 百健艾迪(BiogenIdec)联合艾伯维(AbbVie)公司研发的人源化达珠单抗(daclizumab, Zenapax)上市用于防止肾移植患者环孢霉素和糖皮质激素的急性排斥反应。然而在应用的过程中发现, 至少有10%患者发生失眠、震颤、头痛、动脉高血压、呼吸困难、胃肠道不良反应和水肿。极少数情况下, 药物可导致严重过敏反应[27]。由于缺乏市场需求, 2009年停止Zenapax在肾抑制患者急性排斥反应的应用。2016年, FDA获准在采用新商品名称(Zinbryta)后用于治疗成人复发型多发性硬化症。
2.1.7 SLAMF7 (signaling lymphocytic activation molecule F7)膜蛋白SLAMF7/CD319/CRACC (CD2-like receptor-activating cytotoxic cells)属于信号淋巴细胞激活分子家族成员, 最早研究发现其参与天然杀伤细胞(NK细胞)黏附功能[28]。与正常浆细胞相比, 多种免疫细胞包括NK细胞、NK样T细胞、CD8+ T细胞、活化单核细胞及树突状细胞均低表达SLAMF7。2008年, Tai等[29]研究发现多发性骨髓瘤患者浆细胞高表达SLAMF7。2015年, 施贵宝与艾伯维联合研发的人源化单克隆抗体依托珠单抗(elotuzumab, Empliciti)获准与revlimid/地塞米松联用治疗复发型多发性骨髓瘤。Elotuzumab既可通过ADCC途径靶向肿瘤细胞, 同时也可以激活NK细胞, 最终达到杀死多发性骨髓瘤的目的[29]。目前, 多个临床试验正在评测elotuzumab在多发性骨髓瘤前期治疗、中期维持及疾病复发的治疗效果。最令人期待的Ⅲ期临床试验CheckMate602 (NCT02726581) 中采用PD-1抗体nivolumab与elotuzumab、地塞米松联用治疗复发型多发骨髓瘤[29]。
2.1.8 CD38多种免疫细胞包括CD4+ T细胞、CD8+ T细胞、B细胞和NK细胞表达膜糖蛋白CD38。CD38在细胞黏附和钙信号转导作用中发挥关键作用。CD38催化环腺苷二磷酸核糖(cyclic ADP-ribose, cADPR)合成及降解。cADPR作用于ryanodine受体(RyRs), 参与细胞内钙库的钙动员。研究表明, CD38/cADPR通过RyRs通道的Ca2+释放和介导的Ca2+信号传递, 在Ca2+内平衡的调控中发挥着重要的作用。CD38分子是慢性B淋巴细胞白血病的预测因子, 也是自身免疫反应性糖尿病的诊断指标, 同时还可用于艾滋病和巨细胞病毒的检测及系统性红斑狼疮的病情监测。2015年, 强生公司联合开发的全人源抗CD38抗体达雷木单抗(daratumumab, Darzalex)获准用于曾用3种以上疗法的复发型、耐药型多发性骨髓瘤患者。2016年, 获准联用lenalidomide (或bortezomib)、地塞米松治疗曾用1种以上疗法的多发性骨髓瘤患者。研究发现, daratumumab通过ADCC以及CDC作用获得杀伤肿瘤细胞的目的。
2.2 激活针对肿瘤的免疫固有性和适应性 2.2.1 TNF-αTNF-α是急性炎症反应期释放的细胞因子, 主要通过活化单核巨噬细胞分泌, 在炎症反应调节和细胞存活过程中发挥关键作用。适量TNF-α有助于肿瘤预防和病原菌抵抗, 然而过量TNF-α可能造成多种病理损伤并促进肿瘤发生发展[1]。1998年, FDA获准高特异阻断TNF-α嵌合抗体infliximab (Remicade)用于治疗克罗恩病、溃疡性结肠炎、牛皮癣、银屑病关节炎、强直性脊柱炎及类风湿性关节炎。2003年, 美国雅培公司联合开发的全人源阿达木单抗(adalimumab, Humira)用于治疗类风湿关节炎和强直性脊柱炎。Infliximab和adalimumab均能中和血中游离型TNF-α和免疫细胞表面跨膜型TNF-α活性, 阻断TNF-α与Ⅰ型TNF-α受体p55及Ⅱ型TNF-α受体p75的亚基结合[30]。
2008年, UCB公司研发的赛妥珠单抗(certoli zumab pegol, Cimzia)获批用于克罗恩病、类风湿性关节炎、银屑病关节炎及强直性脊柱炎的治疗。赛妥珠单抗是人源化抗体Fab片段经聚乙二醇化TNF-α抑制剂[31]。2009年, 强生公司联合研发的全人源化戈利木单抗(golimumab, Simponi)获准上市治疗关节炎、银屑病关节炎和强直性脊柱炎等疾病(PMID20065639)。戈利木单抗是经人源TNF-α免疫humab (Medarex)转基因小鼠产生的杂交瘤克隆中分离获得。2016年, 美国获准辉瑞公司的嵌合型单克隆抗体英夫利昔单抗(infliximab-dybb, Inflectra)作为第一个生物类似药上市, 为银屑病性关节炎、强直性脊柱炎、类风湿关节炎和克罗恩病患者提供Remicade以外的选择。
2.2.2 IL-6及IL-6R多功能细胞因子白细胞介素6 (IL-6) 具备促进和抑制炎症反应的双重作用, 在多种疾病发生发展过程中发挥关键作用。组织损伤及细菌感染期间, T细胞和巨噬细胞释放大量IL-6刺激免疫应答[32]。另一方面, IL-6通过抑制TNF-α和IL-1及活化IL-1Ra和IL-10发挥抑制炎症作用。研究发现, IL-6结合IL-6Rα链(CD126), CD126与信号转导组分gp130 (CD130) 共同组成细胞表面I型细胞因子受体复合物通过JAK-STAT信号转导通路激活下游信号[33]。细胞因子IL-6除存在膜结合受体外, 也存在可溶形式的IL-6R (sIL-6R)。研究发现, 神经元细胞虽对IL-6单纯刺激无反应, 但是sIL-6R调控神经突触生长并促进神经元的存活。2010年, 靶向IL-6R的人源化托珠单抗(tocilizumab, 罗氏公司Actemra)获准用于治疗类风湿性关节炎和全身性青少年特发性关节炎[34]。此外, 2014年杨森公司生产的嵌合单克隆抗体西妥昔单抗(siltuximab, Sylvant)靶向细胞因子IL-6治疗艾滋病毒(HIV)和人类疱疹病毒-8 (HHV-8) 等疾病。Siltuximab阻断IL-6结合可溶性及细胞膜型IL-6R, 抑制IL-6介导的B淋巴细胞和浆细胞增殖、VEGF分泌及自身免疫的发生。
2.2.3 IgE过敏反应的关键因子IgE主要通过呼吸道和消化道黏膜固有层淋巴组织B细胞分泌合成。阻断IgE合成与IgE炎性反应通路成为抗过敏及抗哮喘潜在药靶。2004年, 诺华公司研制的人源化抗IgE抗体奥马珠单抗(omalizumab, Xolair)用于治疗哮喘、荨麻疹和湿疹等过敏性疾病[35]。Omalizumab降低血浆游离IgE水平, 阻断IgE与肥大细胞、嗜碱性粒细胞上IgE受体结合获得治疗效果。临床应用过程中发现, 该抗体具有良好安全性和耐受性。
2.2.4 RANKLⅡ型膜蛋白RANKL属于肿瘤坏死因子超家族成员, 在骨骼肌、胸腺、肝、结肠、小肠、肾上腺、成骨细胞、乳腺上皮细胞、前列腺及胰腺均有表达。RANKL参与凋亡, 同时能够通过免疫系统调控骨再生及重塑[36, 37]。2010年, Amgen公司研制的全人单克隆抗体地诺单抗(denosumab, Prolia)能够抑制破骨细胞的活化及发展, 降低骨吸收, 增加骨密度, 用于绝经后骨质疏松症的治疗[38]。较其他RANKL抑制剂, denosumab能够在更低浓度下产生药效, 同时具有作用时间长和无自身免疫排斥反应等特点。临床研究发现, denosumab可在12 h内起效, 半衰期约为32天, 个别患者的药效可长达6个月[39]。
2.2.5 IL-12/IL-23细胞因子IL-12在Th1型细胞介导的炎性免疫反应中发挥关键作用。细胞因子IL-23亦属于IL-12家族, 与IL-12共用p40亚单位发挥作用。自身免疫性疾病斑块型银屑病主要表现为炎症斑块和鳞屑状皮肤, 伴随IL-12和IL-23等细胞因子异常增高。2008年, 强生公司研发靶向p40亚基阻断IL-12/IL-23细胞因子效应的全人单克隆抗体ustekinumab (Stelara)上市, 用于治疗中度至重度斑块性银屑病[40]。2013年增加治疗牛皮藓性关节炎新适应证。2016年底, 强生公司向FDA递交靶向IL-23 p19亚单位单抗guselkumab, 用于治疗中至重度斑块性银屑病, Ⅲ期临床试验结果显示该单抗效果明显优于阿达木单抗。
2.2.6 IL-1βIL-1β调节炎症免疫反应, 过度活化的IL-1β也会引起免疫系统疾病[41]。2009年, 诺华公司靶向IL-1β的全人单克隆抗体药物canakinumab (Ilaris)上市, 用于治疗与cryopyrin蛋白相关的周期综合征(CAPS)。2016年, FDA增加该抗体的3种罕见严重自身炎症疾病适应证, 包括肿瘤坏死因子受体相关周期综合征(TRAPS)、超免疫球蛋白D综合征(HIDS)/甲羟戊酸激酶缺陷(MKD)和地中海热(FMF)。Canakinumab抗体皮下注射后约7天可达最高血药浓度, 绝对生物利用度达到70%。抗体主要通过受体介导的胞饮作用或液相内吞代谢, 半衰期大约26天左右。
2.2.7 IL-5细胞因子IL-5调节嗜酸性粒细胞的生长、活化和存活。IL-5在嗜酸性粒细胞从骨髓迁移至肺部及其他器官时发挥关键作用。2015年, 葛兰素史克(GSK)公司研发的一种靶向IL-5的全人源化单克隆抗体美泊利单抗(mepolizumab, Nucala)获批, 用于治疗严重嗜酸粒细胞性哮喘[42]。Mepolizumab通过直接与细胞因子IL-5结合, 阻断IL-5与嗜酸性粒细胞表面的IL-5受体结合, 降低组织、血液和痰液中的嗜酸性粒细胞水平及其介导的炎性反应。皮下注射mepolizumab的生物利用度可达80%, 大约4~8天达最高血药浓度。2016年, 梯瓦公司研发的靶向IL-5的全人源单抗reslizumab (Cinqair)用于治疗嗜酸性粒细胞哮喘。临床应用发现, reslizumab能够降低严重嗜酸粒细胞性哮喘患者的恶化程度, 同时提高患者肺功能和降低哮喘程度。常见不良反应包括血液肌酸磷酸激酶增加、肌痛及过敏反应。Mepolizumab和reslizumab均通过蛋白水解酶降解代谢, 其生物半衰期分别为20天和24天。
2.2.8 α4β7整合素是淋巴细胞的肠道迁移关键蛋白, 在肠道疾病的发生发展过程中发挥关键作用[43]。2014年, 日本武田(Takeda)公司研发的靶向α4β7人源化单克隆抗体的维多珠单抗(vedolizumab, Entyvio)获准上市用于治疗中到重度传染性法氏囊病(IBD), 后期增加溃疡性结肠炎(UC)和克罗恩病(CD)等新适应证。Vedolizumab通过结合淋巴细胞α4β7整联蛋白, 预防淋巴细胞迁移入肠组织。与靶向α4那他珠单抗不同, vedolizumab不阻止淋巴细胞迁移进入中枢神经系统。因此, 临床应用过程中不会出现乳多空JC病毒介导的进行性多灶性白质脑病。Vedolizumab引起的严重感染比例低, 因此在应用前也不需要对结核病毒和乙型肝炎病毒进行筛查。
2.2.9 BAFFB细胞激活因子(BAFF)在维持B细胞发育和存活过程中发挥关键作用。研究发现, 自身免疫性疾病系统性红斑狼疮(SLE)发生过程中BAFF表达异常增高, 引起自身免疫性B细胞异常增殖, 进而促进SLE发展。2011年, 葛兰素史克公司联合开发的靶向BAFF单克隆抗体药物belimumab (Benlysta)成为56年来第一个用于治疗SLE的药物。Belimumab通过结合BAFF, 防止BAFF激活B细胞, 导致SLE患者B细胞凋亡, 有效阻止SLE患者B细胞造成的自身免疫损伤。
2.3 抗体偶联细胞毒药物(antibody-drug conjugate, ADCs)ADCs是一类将化疗药物与抗体偶联的药物, 能够杀死肿瘤细胞, 并且不良反应较少。Emtansine (DM1) 以及MMAE是常用的两种偶联化疗药物[44, 45]。
2013年, 基因泰克公司研发的ado-trastuzumab emtansine (Kadcyla)通过曲妥珠单抗(赫赛汀)偶联细胞毒性剂DM1靶向HER2。Kadcyla不仅通过曲妥珠单抗阻断HER2/neu受体同二聚化或异二聚化, 抑制其介导的信号传导通路, 同时DM1进入细胞后结合微管蛋白抑制肿瘤细胞生长。Kadcyla主要用于治疗晚期HER2阳性转移性乳腺癌, 常见不良反应包括恶心、乏力、血小板减少和肌肉骨骼酸痛等。临床研究发现, Kadcyla不仅为HER2阳性乳腺癌患者提供更长的无进展生存期, 同时其严重不良反应事件仅为赫赛汀及DM1单用的一半。
抗有丝分裂剂去甲基auristatin E (MMAE)阻断微管蛋白的聚合而抑制细胞分裂。临床无法药用是由于MMAE毒性过强。抗体药物经特殊侧链偶联MMAE结构稳定, 而经细胞内吞后在蛋白酶水解作用下释放MMAE单体, 进而导致肿瘤细胞周期停滞及凋亡[46]。Brentuximab vedotin (Adcetris)是一种偶联抗CD30抗体和MMAE的药物。2011年获批主要用于治疗霍杰金淋巴瘤(HL)和一种罕见的系统性间变性大细胞淋巴瘤(ALCL)。抗肿瘤特异性蛋白的单克隆抗体联合细胞毒药物成为未来ADCs的发展趋势。多项研究发现, 这些具有特异靶向肿瘤细胞的ADCs具有令人期待的治疗效果。例如用于乳腺癌及黑色素瘤的glembatumumab vedotin (anti-HPNMB ADC), 用于慢性淋巴细胞白血病的cituzumab vendotin (anti ROR1 ADC)[47-50]。
3 靶向免疫细胞/免疫节点相关单克隆抗体进展 3.1 靶向免疫抑制分子如CTLA-4、PD-1及PD-L1白细胞分化抗原细胞毒T淋巴细胞相关抗原4 (CTLA-4) 是一种T细胞跨膜受体。CTLA-4与其配基B7分子结合后抑制T细胞免疫活性, 诱导免疫耐受的形成[51]。2011年, 靶向CTLA-4全人单克隆抗体ipilimumab (Yervoy)获批用于黑色素瘤的治疗, 后扩展到转移性肾癌、淋巴瘤、胰腺癌、前列腺癌、肺癌和膀胱癌等肿瘤疾病。Ipilimumab的半衰期约为15天, 常见不良反应主要是T细胞过度活化和增殖引起免疫不良反应, 发生率约为10%~20%[52]。
程序性细胞死亡蛋白1 (PD-1, CD279) 属于免疫球蛋白超家族的细胞膜受体, 主要表达于T细胞及B细胞[53]。PD-1受体存在PD-L1和PD-L2两种配基。PD-1活化T细胞, 与其配基结合抑制T细胞活性。PD-1抑制性免疫信号通过促进淋巴结抗原特异性T细胞凋亡和减少调节性T细胞凋亡实现。研究发现, 靶向PD-1抑制性免疫信号通路成为治疗感染疾病、肿瘤和自身免疫性疾病的潜在药靶。2014年9月, 默沙东研发的人源化单克隆抗体pembrolizumab (Keytruda)成为第一个获准的抗PD1单抗, 主要用于伊匹单抗(ipilimumab, 抗CTLA-4抗体)治疗无效的不可切除性或转移性黑色素瘤患者。单抗通过结合T细胞PD-1, 阻断肿瘤细胞通过PD-L1产生的信号, 重新激活免疫系统对抗肿瘤。2015年ipilimumab获准进行转移性非小细胞肺癌、转移性头颈部鳞癌及霍奇金淋巴瘤治疗。2014年12月, 施贵宝公司靶向PD-1的全人源纳武单抗(nivolumab, Opdivo)上市用于治疗晚期鳞状非小细胞肺癌(NSCLC)患者。Nivolumab主要的不良反应包括肺、结肠、肝、肾及甲状腺的严重免疫炎症反应等。研究发现, 肿瘤细胞PD-L1高表达与肿瘤免疫耐受形成密切相关。肿瘤细胞PD-L1能够结合免疫细胞PD-1和CD80受体(B7-1Rs), 而阻断肿瘤细胞PD-L1能够恢复免疫系统识别并杀伤肿瘤的能力。2016年, 基因泰克公司靶向PD-L1单抗atezolizumab (Tecentriq)获准用于膀胱癌和转移性非小细胞肺癌的治疗, 目前正在进行结肠癌、黑色素瘤、乳腺癌及肾癌的临床试验。临床需要每3周静脉输注1 200 mg单抗, 常见不良反应包括恶心、尿路感染、疲乏及食欲减退等。
3.2 靶向免疫激活分子如CD137/OX40肿瘤坏死因子受体超家族成员4 (TNFRSF4, CD137, OX40) 主要表达在活化T细胞及炎症反应过程的树突细胞、B细胞、滤泡树突细胞、NK细胞、粒细胞及血管壁细胞。CD137在调节肿瘤免疫及T细胞活化、增殖、黏附等方面发挥关键作用[54]。2016年, 辉瑞公司与美国国家癌症研究所(NCI)联合研发全人IgG2单克隆抗体utomilumab。此激活型单抗结合CD137诱导T细胞免疫反应, 与抗PD-1单抗联用产生更强的抗肿瘤效果[55]。
4 其他 4.1 抗IL-17A治疗炎性相关疾病细胞因子IL-17A主要通过T细胞产生, 与其受体IL-17RA、IL-17RF结合后刺激纤维细胞、胆道上皮细胞释放IL-1、IL-6、TNF-α以及CXCL1等促炎因子, 最终导致类风湿性关节炎、牛皮癣和多发性硬化等慢性炎性疾病[56]。2015年, 诺华公司全人源IgG1κ型抗IL-17A抗体苏金单抗(secukinumab, Cosentyx)上市用于治疗中度、重度斑块状银屑病及强直性脊柱炎。2016年, 礼来公司人源化单克隆抗体伊珠单抗(ixekizumab, Talz)成为第二个获准上市的IL-17A单抗, 用于治疗成人中重度斑块状银屑病。2017年2月, Valeant公司研发的抗IL-17A受体单克隆抗体brodalumab (Siliq)获准用于治疗中重度银屑病, 成为第一个受体水平阻断IL-17A信号通路的单抗[57, 58]。3种靶向IL-17A的抗体均可通过皮下注射获得满意的治疗效果。
4.2 炭疽毒素保护抗原(protective antigen of anthrax toxin, PA)罕见疾病吸入性炭疽由吸入炭疽杆菌孢子所致, 主要通过空气释放扩散。当人体接触到受感染的动物、或受污染的动物产品或人为蓄意释放(生化袭击)的炭疽孢子后引发感染。炭疽杆菌能在体内增殖并产生毒素, 引发严重不可逆的组织损伤并导致死亡[59]。2001年炭疽杆菌恐怖袭击造成5人死亡事件, 不仅发现炭疽毒素对多种抗生素耐受, 同时发现对抗或者中和毒素可能是新的有效治疗手段。2012年, FDA获准美国人类基因组科学公司(Human Genome Sciences, HGS)研制的靶向PA人源化瑞西巴库单抗(raxibacumab)治疗吸入性炭疽。Raxibacumab主要通过皮下注射, 不能用于静脉输注或皮内注射[60]。2016年美国Elusys Therapeutics公司研制嵌合单克隆抗体obiltoxaximab (Anthim)也获准作为吸入性炭疽的治疗及预防性药物。静脉注射obiltoxaximab能够在获得性免疫系统激活前提供大约2~3周左右的PA中和效应[61]。
4.3 达比加群酯(diabigatran)Diabigatran是新一代口服抗凝药物直接凝血酶抑制剂, 2010年获批用于预防非瓣膜性房颤患者的卒中和全身性栓塞, 目前此适应证在全球60多个国家获准。Diabigatran提供的抗凝效果稳定且有效, 同时极少会有药物相互作用的发生, 无需进行常规的凝血功能监测或剂量调整。2015年, 勃林格殷格翰公司研制的具有拮抗diabigatran效应的单克隆抗体idarucizumab (Praxbind)获批。此抗体可以在数分钟内逆转diabigatran的抗凝作用, 有效防止diabigatran使用者因偶然出血造成的致命伤害[62]。
4.4 Proprotein convertase subtilisin/kexin type 9 (PCSK9)最近研究发现, PCSK9在维持脂蛋白内稳态过程中发挥关键作用。PCSK9在肝细胞表面结合低密度脂蛋白受体(LDLR)并介导LDLR的降解。由于LDLR是LDL清除的主要受体, 所以异常增高的PCSK9不仅导致LDLR水平降低, 同时引起LDL-C水平增高。功能获得型和功能缺失型PCSK9突变分别导致高胆固醇血症和低胆固醇血症[63]。2015年, 赛诺菲公司联合开发全人单克隆阿利库单抗(alirocumab, Praluent)及Amgen生物制药公司研发的全人单克隆抗体依洛珠单抗(evolocumab, Repatha)获准治疗动脉粥样硬化性心血管疾病或杂合子家族性高胆固醇血症。抗PCSK9单抗均可通过皮下给药, 抑制PCSK9与LDLR结合, 进而提高LDLR表达并降低LDL-C水平。
5 总结与展望在过去30年间, FDA批准了多种用于疾病治疗和诊断的单克隆抗体药物[1]。单克隆抗体药物在自身免疫病和肿瘤治疗领域获得巨大进展, 同时也是医药领域增长速度最快、最有前景的发展方向。目前用于治疗风湿性关节炎的单抗药物占据约13%的市场份额。单克隆抗体药物在血液病(如慢性淋巴细胞白血病和霍奇金淋巴瘤等)、实体瘤(如鳞状细胞癌等)、原发性肿瘤(如多形性成胶质细胞瘤等)和肿瘤转移(如骨转移)治疗过程中发挥重要作用。同时单克隆抗体药物也展示出对呼吸系统疾病(如过敏性哮喘)、中枢神经系统退行性疾病(如多发性硬化)及慢性胃肠道病症(如克罗恩病)治疗前景[12]。截至2016年12月, 还有20个用于肿瘤适应证及32个用于非肿瘤适应证的单克隆抗体正在进行Ⅲ期或Ⅱ/Ⅲ期临床试验。这些抗体用于获得性免疫缺陷综合征、获得性血栓性血小板减少性紫癜、阿尔茨海默病、偏头痛、系统性红斑狼疮及肿瘤治疗[3]。
尽管早期的单克隆抗体药物存在缺陷, 但是研究者始终没有放弃对单抗改造的研究[12, 64]。单克隆抗体可以通过进一步的基因工程改造提高亲和力、稳定性和生物学活性。例如, 研究发现IgG亚类和特异性Fc受体之间的相互作用存在显著差异; 抗体糖基化或其他翻译后修饰改造可影响抗体特异性、生物学活性及半衰期[65]。这些生物技术的提高会不断推进单克隆抗体药物的发展。毫无疑问, 单克隆抗体药物改变了制药工业和医学实践, 并为常规治疗无效的患者提供新的希望[66]。在未来, 单克隆抗体依然将作为一种最为有效治疗手段用于疾病的诊断、治疗及研究[1, 12]。
| [1] | Buss NA, Henderson SJ, McFarlane M, et al. Monoclonal antibody therapeutics: history and future[J]. Curr Opin Pharmacol, 2012, 12: 615–622. DOI:10.1016/j.coph.2012.08.001 |
| [2] | Weiner GJ. Building better monoclonal antibody-based therapeutics[J]. Nat Rev Cancer, 2015, 15: 361–370. DOI:10.1038/nrc3930 |
| [3] | Reichert JM. Antibodies to watch in 2017[J]. MAbs, 2017, 9: 167–181. DOI:10.1080/19420862.2016.1269580 |
| [4] | Reichert JM. Antibodies to watch in 2016[J]. MAbs, 2016, 8: 197–204. DOI:10.1080/19420862.2015.1125583 |
| [5] | Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity[J]. Nature, 1975, 256: 495–497. DOI:10.1038/256495a0 |
| [6] | Ober RJ, Radu CG, Ghetie V, et al. Differences in promiscuity for antibody-FcRn interactions across species: implications for therapeutic antibodies[J]. Int Immunol, 2001, 13: 1551–1559. DOI:10.1093/intimm/13.12.1551 |
| [7] | Stern M, Herrmann R. Overview of monoclonal antibodies in cancer therapy: present and promise[J]. Crit Rev Oncol Hematol, 2005, 54: 11–29. DOI:10.1016/j.critrevonc.2004.10.011 |
| [8] | Morrison SL, Johnson MJ, Herzenberg LA, et al. Chimeric human antibody molecules: mouse antigen-binding domains with human constant region domains[J]. Proc Nat Acad Sci U S A, 1984, 81: 6851–6855. DOI:10.1073/pnas.81.21.6851 |
| [9] | Presta LG. Engineering of therapeutic antibodies to minimize immunogenicity and optimize function[J]. Adv Drug Deliv Rev, 2006, 58: 640–656. DOI:10.1016/j.addr.2006.01.026 |
| [10] | Jones PT, Dear PH, Foote J, et al. Replacing the complementarity-determining regions in a human antibody with those from a mouse[J]. Nature, 1986, 321: 522–525. DOI:10.1038/321522a0 |
| [11] | Baker MP, Reynolds HM, Lumicisi B, et al. Immunogenicity of protein therapeutics: the key causes, consequences and challenges[J]. Self Nonself, 2010, 1: 314–322. DOI:10.4161/self.1.4.13904 |
| [12] | Rodgers KR, Chou RC. Therapeutic monoclonal antibodies and derivatives: historical perspectives and future directions[J]. Biotechnol Adv, 2016, 34: 1149–1158. DOI:10.1016/j.biotechadv.2016.07.004 |
| [13] | Mitri Z, Constantine T, O'Regan R. The HER2 receptor in breast cancer: pathophysiology, clinical use, and new advances in therapy[J]. Chemother Res Pract, 2012, 2012: 743193. |
| [14] | Shaw T, Quan J, Totoritis MC. B cell therapy for rheumatoid arthritis: the rituximab (anti-CD20) experience[J]. Ann Rheum Dis, 2003, 62(Suppl 2): ii55–ii59. |
| [15] | Rizzieri D. Zevalin® (ibritumomab tiuxetan): after more than a decade of treatment experience, what have we learned?[J]. Crit Rev Oncol Hematol, 2016, 105: 5–17. DOI:10.1016/j.critrevonc.2016.07.008 |
| [16] | Dhillon S. Obinutuzumab: a review in rituximab-refractory or-relapsed follicular lymphoma[J]. Target Oncol, 2017, 12: 255–262. DOI:10.1007/s11523-017-0485-6 |
| [17] | Ratner M. Genentech's glyco-engineered antibody to succeed Rituxan[J]. Nat Biotechnol, 2014, 32: 6–7. |
| [18] | Umana P, Jean-Mairet J, Moudry R, et al. Engineered glycoforms of an antineuroblastoma IgG1 with optimized antibody-dependent cellular cytotoxic activity[J]. Nat Biotechnol, 1999, 17: 176–180. DOI:10.1038/6179 |
| [19] | Evans SS, Clemmons AB. Obinutuzumab: a novel anti-CD20 monoclonal antibody for chronic lymphocytic leukemia[J]. J Adv Pract Oncol, 2015, 6: 370–374. |
| [20] | Rimawi MF, Schiff R, Osborne CK. Targeting HER2 for the treatment of breast cancer[J]. Ann Rev Med, 2015, 66: 111–128. DOI:10.1146/annurev-med-042513-015127 |
| [21] | Chang L, Li CH, Gao J. Progress in the study of Her2-targeted cancer therapeutic antibodies[J]. Acta Pharm Sin (药学学报), 2015, 50: 516–520. |
| [22] | Goel HL, Mercurio AM. VEGF targets the tumour cell[J]. Nat Rev Cancer, 2013, 13: 871–882. DOI:10.1038/nrc3627 |
| [23] | Tebbutt N, Pedersen MW, Johns TG. Targeting the ERBB family in cancer: couples therapy[J]. Nat Rev Cancer, 2013, 13: 663–673. DOI:10.1038/nrc3559 |
| [24] | Katz BZ, Herishanu Y. Therapeutic targeting of CD19 in hematological malignancies: past, present, future and beyond[J]. Leuk Lymphoma, 2014, 55: 999–1006. DOI:10.3109/10428194.2013.828354 |
| [25] | Molhoj M, Crommer S, Brischwein K, et al. CD19-/CD3-bispecific antibody of the BiTE class is far superior to tandem diabody with respect to redirected tumor cell lysis[J]. Mol Immunol, 2007, 44: 1935–1943. DOI:10.1016/j.molimm.2006.09.032 |
| [26] | Triplett TA, Curti BD, Bonafede PR, et al. Defining a functionally distinct subset of human memory CD4+ T cells that are CD25POS and FOXP3NEG[J]. Eur J Immunol, 2012, 42: 1893–1905. DOI:10.1002/eji.v42.7 |
| [27] | European Medicines Agency. European public assessment reprot for Zenapax [R]. London, EMEA, 2007. |
| [28] | Friend R, Bhutani M, Voorhees PM, et al. Clinical potential of SLAMF7 antibodies-focus on elotuzumab in multiple myeloma[J]. Drug Des Devel Ther, 2017, 11: 893–900. DOI:10.2147/DDDT |
| [29] | Tai YT, Dillon M, Song W, et al. Anti-CS1 humanized monoclonal antibody HuLuc63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow milieu[J]. Blood, 2008, 112: 1329–1337. DOI:10.1182/blood-2007-08-107292 |
| [30] | Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis[J]. New Engl J Med, 2001, 344: 907–916. DOI:10.1056/NEJM200103223441207 |
| [31] | Deeks ED. Certolizumab pegol: a review in inflammatory autoimmune diseases[J]. BioDrugs, 2016, 30: 607–617. DOI:10.1007/s40259-016-0197-y |
| [32] | van der Poll T, Keogh CV, Guirao X, et al. Interleukin-6 gene-deficient mice show impaired defense against pneumococcal pneumonia[J]. J Infect Dis, 1997, 176: 439–444. DOI:10.1086/jid.1997.176.issue-2 |
| [33] | Heinrich PC, Behrmann I, Muller-Newen G, et al. Interleukin 6-type cytokine signalling through the gp130/Jak/STAT pathway[J]. Biochem J, 1998, 334: 297–314. DOI:10.1042/bj3340297 |
| [34] | Kallen KJ. The role of transsignalling via the agonistic soluble IL-6 receptor in human diseases[J]. Biochim Biophys Acta, 2002, 1592: 323–343. DOI:10.1016/S0167-4889(02)00325-7 |
| [35] | Luu M, Bardou M, Bonniaud P, et al. Pharmacokinetics, pharmacodynamics and clinical efficacy of omalizumab for the treatment of asthma[J]. Exp Opin Drug Metab Toxicol, 2016, 12: 1503–1511. DOI:10.1080/17425255.2016.1248403 |
| [36] | Mueller CG, Hess E. Emerging functions of RANKL in lymphoid tissues[J]. Front Immunol, 2012, 3: 261. |
| [37] | Wada T, Nakashima T, Hiroshi N, et al. RANKL-RANK signaling in osteoclastogenesis and bone disease[J]. Trend Mol Med, 2006, 12: 17–25. DOI:10.1016/j.molmed.2005.11.007 |
| [38] | Zaheer S, LeBoff M, Lewiecki EM. Denosumab for the treatment of osteoporosis[J]. Exp Opin Drug Metab Toxicol, 2015, 11: 461–470. DOI:10.1517/17425255.2015.1000860 |
| [39] | Body JJ, Facon T, Coleman RE, et al. A study of the biological receptor activator of nuclear factor-κB ligand inhibitor, denosumab, in patients with multiple myeloma or bone metastases from breast cancer[J]. Clin Cancer Res, 2006, 12: 1221–1228. DOI:10.1158/1078-0432.CCR-05-1933 |
| [40] | Yeilding N, Szapary P, Brodmerkel C, et al. Development of the IL-12/23 antagonist ustekinumab in psoriasis: past, present, and future perspectives--an update[J]. Ann N Y Acad Sci, 2012, 1263: 1–12. DOI:10.1111/nyas.2012.1263.issue-1 |
| [41] | Satoh T, Otsuka A, Contassot E, et al. The inflammasome and IL-1beta: implications for the treatment of inflammatory diseases[J]. Immunotherapy, 2015, 7: 243–254. DOI:10.2217/imt.14.106 |
| [42] | Walsh GM. Mepolizumab-based therapy in asthma: an update[J]. Curr Opin Allergy Clin Immunol, 2015, 15: 392–396. DOI:10.1097/ACI.0000000000000183 |
| [43] | Ley K, Rivera-Nieves J, Sandborn WJ, et al. Integrin-based therapeutics: biological basis, clinical use and new drugs[J]. Nat Rev Drug discov, 2016, 15: 173–183. DOI:10.1038/nrd.2015.10 |
| [44] | Tsuchikama K, An Z. Antibody-drug conjugates: recent advances in conjugation and linker chemistries[J]. Protein Cell, 2016. DOI:10.1007/s13238-016-0323-0 |
| [45] | Guo JJ, Gao R, Quan TF, et al. Progress on pharmacokinetic study of antibody-drug conjugates[J]. Acta Pharm Sin (药学学报), 2015, 50: 1203–1209. |
| [46] | Francisco JA, Cerveny CG, Meyer DL, et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity[J]. Blood, 2003, 102: 1458–1465. DOI:10.1182/blood-2003-01-0039 |
| [47] | Yardley DA, Weaver R, Melisoko ME, et al. EMERGE: a randomized phase Ⅱ study of the antibody-drug conjugate glembatumumab vedotin in advanced glycoprotrin NMB-expressing breast cancer[J]. J Clin Oncol, 2015, 33: 1609–1619. DOI:10.1200/JCO.2014.56.2959 |
| [48] | Widhopf GF 2nd, Cui B, Ghia EM, et al. ROR1 can interact with TCL1 and enhance leukemogenesis in Eu-TCL1 transgenic mice[J]. Proc Natl Acad Sci U S A, 2014, 111: 793–798. DOI:10.1073/pnas.1308374111 |
| [49] | Cui B, Zhang S, Chen L, et al. Targeting ROR1 inhibits epithelial-mesenchymal transition and metastasis[J]. Cancer Res, 2013, 73: 3649–3660. DOI:10.1158/0008-5472.CAN-12-3832 |
| [50] | Cui B, Widhopf GF 2nd, Prussak CE, et al. Cirtuzumab vedotin (UC-961ADC3), an anti-ROR1 monomethyl auristatin E antibody-drug conjugate, is a potential treatment for ROR1-positive leukemia and solid tumors[J]. Blood, 2013, 122: 1637. |
| [51] | Walker LS, Sansom DM. Confusing signals: recent progress in CTLA-4 biology[J]. Trend Immunol, 2015, 36: 63–70. DOI:10.1016/j.it.2014.12.001 |
| [52] | Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study[J]. Lancet Oncol, 2010, 11: 155–164. DOI:10.1016/S1470-2045(09)70334-1 |
| [53] | Topalian SL, Taube JM, Anders RA, et al. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy[J]. Nat Rev Cancer, 2016, 16: 275–287. DOI:10.1038/nrc.2016.36 |
| [54] | Aspeslagh S, Postel-Vinay S, Rusakiewicz S, et al. Rationale for anti-OX40 cancer immunotherapy[J]. Eur J Cancer, 2016, 52: 50–66. DOI:10.1016/j.ejca.2015.08.021 |
| [55] | An on. Enhancing PD-1 blockade in solid tumors[J]. Cancer Discov, 2016, 6: OF2. |
| [56] | Blauvelt A, Lebwohl MG, Bissonnette R. IL-23/IL-17A dysfunction phenotypes inform possible clinical effects from anti-IL-17A therapies[J]. J Invest Dermatol, 2015, 135: 1946–1953. DOI:10.1038/jid.2015.144 |
| [57] | Puig L. The role of IL 23 in the treatment of psoriasis[J]. Exp Rev Clin Immunol, 2017. DOI:10.1080/1744666X.2017.1292137 |
| [58] | Dong J, Goldenberg G. New biologics in psoriasis: an update on IL-23 and IL-17 inhibitors[J]. Cutis, 2017, 99: 123–127. |
| [59] | Kummerfeldt CE. Raxibacumab: potential role in the treatment of inhalational anthrax[J]. Infect Drug Resist, 2014, 7: 101–109. |
| [60] | Goldenberg MM. Pharmaceutical approval update[J]. P T, 2013, 38: 86–95. |
| [61] | Nagy CF, Mondick J, Serbina N, et al. Animal-to-human dose translation of obiltoxaximab for treatment of inhalational anthrax under the US FDA animal rule[J]. Clin Transl Sci, 2017, 10: 12–19. DOI:10.1111/cts.2017.10.issue-1 |
| [62] | Samos M, Stanciakova L, Skornova I, et al. Review of the pharmacology of the emerging possibilities of the direct oral anticoagulants' reversal[J]. Curr Drug Metab, 2017. DOI:10.2174/1389200218666170413155351 |
| [63] | McKenney JM. Understanding PCSK9 and anti-PCSK9 therapies[J]. J Clin Lipidol, 2015, 9: 170–186. DOI:10.1016/j.jacl.2015.01.001 |
| [64] | Gunn BM, Alter G. Modulating antibody functionality in infectious disease and vaccination[J]. Trend Mol Med, 2016, 22: 969–982. DOI:10.1016/j.molmed.2016.09.002 |
| [65] | Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions[J]. Front Immunol, 2014, 5: 520. |
| [66] | Udpa N, Million RP. Monoclonal antibody biosimilars[J]. Nat Rev Drug Discov, 2016, 15: 13–14. |