 2017, Vol. 52
2017, Vol. 52



人体内的酪氨酸磷酸化水平是由蛋白酪氨酸激酶 (protein tyrosine kinase, PTK) 和蛋白酪氨酸磷酸酶 (protein tyrosine phosphatases, PTPs) 精准调控的。PTK-PTP平衡紊乱会导致酪氨酸磷酸化异常进而引起多种人类疾病, 包括癌症、代谢综合征和自身免疫疾病等[1]。PTP是由多个亚型构成的磷酸酶家族, 主要包括经典的受体PTP (RPTPs)、经典的非受体PTP (NRPTPs)、双特异性PTP (dsPTPs) 和低分子量PTP (LMW-PTPs)[2]四类。其中经典的RPTP和NRPTP是主要的成员。RPTP是细胞膜上的信号转导调节子, 能够在细胞的生长增殖、神经发育和免疫应答中发挥作用[3]; NRPTP成员都具有磷酸酶催化区 (PTP结构域) 以调控SRC家族蛋白激酶 (Src family kinase, SFK) 的磷酸化水平[4, 5], 很多信号通路的调节都与NRPTP成员有关, 例如蛋白酪氨酸磷酸酶1B (protein tyrosine phosphatases 1B, PTP1B) 是胰岛素信号和瘦素信号的调控因子; 含有Src同源2结构域的磷酸酶2 (Src homology-2 domain-containing phosphatase 2, SHP2) 对生长因子和细胞因子的激活有重要作用[1]。淋巴特异性酪氨酸磷酸酶 (lymphoid-specific tyrosine phos phatase, LYP) 作为NRPTP的代表成员, 是T细胞免疫系统的负调控因子, 因其与多种自身免疫病的密切关系而受到越来越多的关注。
1 LYP的结构及功能LYP致病突变最早于2005年被南加州大学的Bottini课题组[6]报道, 与其他PTP成员不同的是, 其特异性地在免疫细胞中表达, 并且需要与C末端SRC激酶 (C-terminal SRC kinase, CSK) 共同发挥作用。该酶可催化LCK、ZAP-70和FYN蛋白的酪氨酸残基去磷酸化, 从而抑制其功能并负向调控T细胞信号通路。LYP可分为C端、N端两个部分。其C端包括200个氨基酸和4个脯氨酸富集区P1区、P2区、P3区和P4区[7], 而多数NRPTP基本不含有脯氨酸富集区或只含有少量脯氨酸富集区。LYP的N端为含有300个氨基酸的磷酸酶催化区 (PTP domain), 该区域是由α螺旋和β折叠构成的, 可分为PTP底物识别区 (H226CSAGCGR233, 包括一个P-loop区)、LYP特异性插入区 (S35TKYKADK42) 和WPD loop区 (残基193-204)。PTP识别区是LYP对底物去磷酸化的功能区; LYP特异性插入区是该酶特有的序列, 也是该酶在结构上与其他NRPTP最大的区别之处, 因此与LYP的底物特异性有关, 且该区域在不同晶体结构中能够呈现出α-螺旋和Loop两种不同二级结构; WPD loop中的Asp195残基为LYP催化底物去磷酸化所必需, 且WPD loop的构象也会影响LYP的活性变化 (图 1)[8]。

|
Figure 1 The structure of protein tyrosine phosphatase (PTP) domain. LYP: Lymphoid-specific tyrosine phosphatase |
在正常生理条件下LYP的脯氨酸富集区与CSK的SH3结构域结合形成复合物, 在T细胞受体信号通路 (T-cell receptor, TCR) 起负调控作用, 通过对关键细胞因子LCK、FYN和ZAP-70去磷酸化, LYP能够抑制下游信号传导从而下调TCR通路, 而CSK则能够磷酸化LCK以适当抵消LYP的抑制作用[1] (图 2)。

|
Figure 2 Function of LYP in TCR signaling. TCR: T-cell receptor; CSK: C-terminal SRC kinase |
许多自身免疫病患者的体内都发现了C1858T突变的PTPn22基因, 研究表明该突变基因会导致LYP620位的精氨酸转变为色氨酸 (R620W)。由于620位残基位于P1区, 其突变会改变P1区的结构, 导致LYP*W620不能够与CSK的SH3结构域结合[7]。大多数观点认为这是一种功能获得性 (gain-of-function) 突变, 可引起LYP功能增强而使TCR信号通路受阻。人体T细胞免疫因而不能及时清除过敏的T细胞, 亢进的LYP同时会上调CD28促进炎症因子的释放[7]而引发自身免疫病[6]。有些不同的观点认为这是一种功能缺失型 (loss-of-function) 突变, LYP不能与CSK结合而失去抑制TCR的作用, 导致不可控的TCR信号传导和异常T细胞激活进而引起自身免疫病[9], 包括Ⅰ型糖尿病、风湿性关节炎、格雷夫斯病以及系统性红斑狼疮等疾病[10]。还有一些研究发现LYP还会通过抑制B细胞受体 (B-cell receptor, BCR) 信号通路, 改变BCR阈值, 提高B细胞敏感性破坏免疫作用的平衡[11]。LYP诱发疾病的机制至今尚不明确, 但可以肯定的是, LYP与免疫功能失调密切相关, 开发小分子LYP抑制剂对于探究LYP的致病机制和治疗自身免疫病有着重大意义。
2 LYP抑制剂近年来发现小分子LYP抑制剂越来越受到关注, 随着计算机辅助药物设计, 高通量筛选等发现先导化合物新技术的迅猛发展和LYP研究的不断深入, 科研人员发现了大量具有良好活性和选择性的小分子抑制剂, 根据其结构和作用方式不同可分为羧酸 (酯) 及抑制剂、非羧酸类抑制剂、金属类抑制剂和共价不可逆抑制剂。
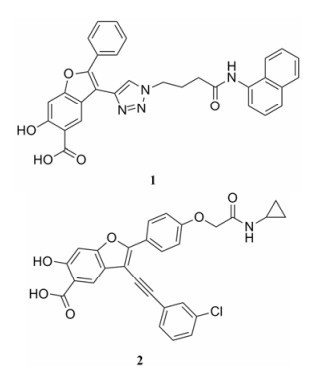
2.1 羧酸 (酯) 类抑制剂 2.1.1 水杨酸 (酯) 类2007年美国印第安纳大学张仲寅教授课题组[12]发现呋喃并水杨酸化合物具有较好的抑制LYP活性, 经过结构优化得到选择性LYP抑制剂I-C11 (1)。该化合物不仅具有较好的抑制活性 (Ki = 2.9 μmol·L-1), 而且细胞通透性良好。2013年, 该课题组通过研究1与LYP的共晶结构发现[10], 该化合物的呋喃并水杨酸母核能够通过氢键、π-π相互作用和静电相互作用与靶酶P-loop区结合, 而萘环则作用于LYP和PTP1B共有活性位点, 因此1对LYP和PTP1B的选择性较差。考虑到先导物结构中侧链苯环和三氮唑并非与靶点结合的关键部位, 因此对该部分进行改造和活性筛选, 得到了抑酶活性和选择性更佳的抑制剂2 (Ki = 0.11 μmol·L-1)。共晶结构显示2的水杨酸母核能够更好地深入到P-loop, 取代苯环的引入增强与PTP识别区的作用, 使得化合物的活性得到增强, 同时由于改造了1的萘环侧链, 规避了与LYP和PTP1B共有活性位点的作用, 提高了化合物2对LYP和PTP1B的选择性。
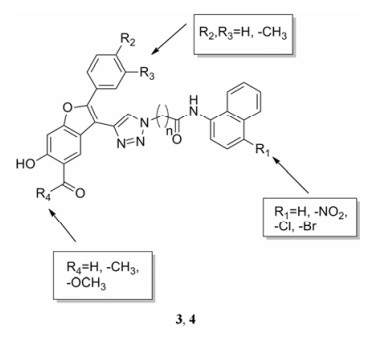
2011年美国Burnham研究院的Lutz Tautz课题组[13]根据化合物1的结构, 应用点击化学方法构建针对LYP靶酶的化合物库, 经活性测试发现化合物3 (R1 = Br, R2 =-CH3, R3 = H, R4 = H, n = 3) 活性最好。体外构效关系研究表明, 当R1为卤素、R2为甲基或甲氧基时活性较好, 但萘环被替换后并未提高活性, 碳链数n = 3时活性最佳。随后选取体外活性较好的化合物进行细胞水平的评价, 发现抑酶活性最好的化合物3不是通过调控TCR通路发挥作用的, 因此未进行深入活性评价。而呋喃并水杨酸甲酯化合物4 (R1 = NO2, R2 =-CH3, R3 = H, R4 =-OCH3, n = 3) 在细胞水平评价中活性最好, 推测其原因可能是4进入细胞后能水解成为活性更强的水杨酸化合物。
|
|
此外, 通过引入酪氨酸磷酸模拟物, 2008年美国哥伦比亚大学的Landry课题组[14]设计了一类噻唑烷酮水杨酸酯类化合物, 活性研究发现化合物5抑制LYP活性最强, IC50值达到0.39 μmol·L-1。此类结构虽然有一定活性, 但属于PAINS化合物[15], 其母核可能具有多种靶点的结合能力, 未来研究还需要进行更多的结构改造。

|
2009年Burnham研究院的Lutz Tautz课题组[16]应用虚拟筛选方法研究了对LYP有一定抑制作用的苗头化合物, 研究策略考虑了WPD loop闭合的活性构象和WPD loop开放的非活性构象, 虚拟筛选后选择71个打分较高的化合物进行活性评价, 进一步活性评价确证了6个活性较好的化合物, 其中化合物6和7的活性最好 (IC50 = 0.947和0.996 μmol·L-1)。分子对接实验结果显示, 在开放的非活性构象中6能够占据催化区外侧的WPD loop, 这种作用效果很强并且具有特异性, 因此利用LYP开放的非活性构象进行虚拟筛选可以得到活性和选择性理想的抑制剂。
该课题组[17]随后于2012年应用高通量筛选方法, 从50 000个化合物中筛选得到715个有初步活性的苗头化合物, 其中190个化合物显示抑制LYP的作用具有剂量依赖性。通过亚型选择性实验进一步得到了33个活性化合物, 经过体内外一系列生物活性评价及构效关系研究得到了先导化合物8 (R1 = 2-CH3, R2 = H, R3 = 3-COOH, IC50 = 0.508 μmol·L-1)。该化合物不仅抑酶活性高而且对同家族结构类似的靶酶具有良好的选择性。例如, 该化合物抑制LYP的活性较PTP1B (IC50 = 1.59 μmol·L-1) 高3倍。细胞活性评价显示, 8能够显著抑制LYP造成的下游信号因子LCK的去磷酸化作用, 从而上调TCR通路信号; 同时该化合物的使用并未引起同家族磷酸酶TCPTP和SHP-1介导的TCR信号衰减, 显示了较好的体内选择性。构效关系研究表明:苯环R1的取代方式对目标结构活性影响较大。该化合物不仅推动了以LYP为靶点治疗自身免疫病的发展, 同时也为研究LYP致病机制提供了新的小分子探针。
化合物9是作者所在课题组[18]基于化合物1和2的晶体结构, 构建基于小分子-靶点相互作用的药效团模型 (interaction-based pharmacophore) 并通过多种虚拟筛选策略和生物活性筛选获得。初步活性表明该化合物具有良好的LYP抑制活性 (IC50 = 6.1 μmol·L-1) 以及选择性; 在人体Jurkat T抗原T细胞中的活性评价实验表明, 9促进ERK和LCK相应酪氨酸残基磷酸化的能力是阳性对照组的两倍; 在加入9的T细胞中, TCR信号介导的细胞因子Nfat/AP-1的表达显著增强 (EC50 = 6.49 μmol·L-1), 以上实验数据表明该化合物能够在细胞水平通过直接抑制LYP影响TCR下游信号通路。从结构角度分析, 9的结构骨架新颖, 不属于PAINS骨架; 而前文提到的化合物8含有硫代巴比妥, 在药物发现研究中一般被认为是PAINS骨架, 易产生脱靶效应 (off-target)。
2.2 非羧酸类抑制剂2011年, 南加州大学的Bottini课题组[19]以对硝基苯磷酸为底物应用高通量筛选从4 000个类药结构中得到了137个苗头化合物, 剔除从结构上推测以氧化作用方式抑制LYP的化合物, 并进行进一步选择性评价, 细胞内生物活性评价得到了四氮唑类先导化合物10。动力学研究表明, 该类结构为非竞争性抑制剂且不含有苯甲酸类磷酸模拟基团。初步构效关系研究表明, 对于R1, 改变环戊基对活性和亚型选择性均有影响, 但对细胞通透性则无影响; 对于R2, 引入带有两个苯环结构的氨基有助于提高活性, 但引入其他基团如哌啶、哌嗪等会使活性降低, 而引入螺环则会增强化合物细胞通透性和相对于HePTP的选择性 (表 1), 根据以上结果进行进一步构效关系研究得到了新型喹啉类抑制剂11, 对接结果显示, 该化合物的四氮唑基团能够与PTP结构域进行氢键作用, 苯环则参与疏水作用。10与11的作用方式基本一致, 通过研究二者对Leu29Ala突变型LYP抑制活性的研究发现, 11活性的提高与其能够与Leu29作用有关。
| Table 1 Structures and activity research of 10 and 11 |






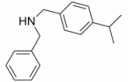
由于在体内蛋白质的构象是在不断变化的, 因此使用单一构象进行虚拟筛选的误差很大, 为了发现新型非羧酸类抑制剂, 作者所在课题组[20]在数据库中剔除羧酸类化合物构建非羧酸化合物库, 应用已知的LYP蛋白-小分子晶体结构复合物 (PDB: 2P6X, 2QCJ和3BRH) 进行筛选得到了23个苗头化合物, 通过体外抑制活性和选择性实验发现了一个新的非苯甲酸类抑制剂化合物12 (IC50 = 7.95 μmol·L-1), 细胞生物活性评价显示12能够磷酸化ERK上调TCR信号通路并具有良好的细胞通透性, 12的发现不仅为开发LYP抑制剂提供了新的方向, 也首创了应用多种晶型进行虚拟筛选发现LYP抑制剂的方法。

|

|
基于之前对金化合物抑制PTP活性的研究[21], 拉荷亚研究所的Brrios课题组[22]发现的金复合物抑制剂13在体内外都显示了良好的活性 (IC50 = 1.5 μmol·L-1) 和选择性。该课题组后续的研究[23]又发现了一个能够选择性抑制单一突变而非双重突变LYP的抑制剂14 (IC50 = 3.5 μmol·L-1)。

|
2014年, 作者所在课题组[18]通过基于相互作用的药效团模型筛选化合物库, 应用优化的打分方法和虚拟筛选得到了29个化合物, 在体外生物活性评价过程中发现了一个新型不可逆抑制剂15 (IC50 = 40.98 μmol·L-1)。拉荷亚研究所的Brrios课题组[24]于2013年发现了一种二硫化物伪不可逆抑制剂16, 在谷胱甘肽条件下可以非竞争性抑制LYP (Ki = 1.1 μmol·L-1), 在二硫苏糖醇中其不可逆抑制作用则部分削弱; 2014年, 该课题组[25]应用高通量筛选发现了一个LYP抑制剂17, 具有良好的活性 (Ki = 0.05 μmol·L-1)。动力学研究表明, 该化合物属于LYP的不可逆抑制剂。由于17的苯甲酸基团属于磷酸模拟基团, 作者推测该基团首先与LYP的活性位点进行结合, 随后化合物17结构中的Michacel受体 (丙烯腈) 可能与靶酶催化位点以外的亲核试剂 (如半胱氨酸残基) 发生反应, 产生了不可逆抑制作用[26, 27]。

|
综上所述, LYP作为自身免疫性疾病的新型靶点受到广泛关注, 近年来研究人员发现了多种结构类型不同的抑制剂。羧酸 (酯) 类抑制剂都能够模拟底物中的磷酸根, 通过羧基与LYP的PTP结构域催化位点 (P-loop) 中的氨基酸残基发生相互作用, 属于竞争性抑制剂。同时, 通过引入其他侧链基团, 不仅提高该类抑制剂对LYP的活性, 而且能与活性位点之外的LYP特异性口袋结合, 从而对其他PTP家族蛋白实现一定的选择性。该类抑制剂普遍具有良好的抑酶活性, 但由于不同PTP家族蛋白的催化位点具有高度的保守性, 因此其选择性的提高也存在较大难度, 较强的负电性也会影响化合物的成药性。而非羧酸类抑制剂中的四氮唑类先导物10是首次发现的非竞争抑制剂, 目前的研究推测可能存在变构调控位点。值得一提的是, 针对其他PTPs (例如PTP1B) 的变构抑制剂已有较多研究报道, 但化合物10和LYP的具体结合方式仍不清楚, 有待进一步研究。非羧酸类抑制剂尽管目前活性还有待提高, 但由于不含羧基有利于提高选择性和成药性, 因此更有发展前途。此外, 金属复合物类抑制剂作为一类新型活性良好的抑制剂同样受到了一定的关注, 但重金属在人体内容易产生毒性, 这为化合物后续的开发造成了一定的困难。而目前发现的不可逆抑制剂一般含有Michacel受体或二硫键, 可能存在脱靶效应, 其研究能否再深入推进, 还有待观察。
| [1] | He RJ, Yu ZH, Zhang RY, et al. Protein tyrosine phosphatases as potential therapeutic targets[J]. Acta Pharmacol Sin, 2014, 35: 1227–1246. DOI:10.1038/aps.2014.80 |
| [2] | Stoker AW. Protein tyrosine phosphatases and signaling[J]. J Endocrinol, 2005, 185: 19–33. DOI:10.1677/joe.1.06069 |
| [3] | Coles CH, Jones EY, Aricescu AR. Extracellular regulation of type Ⅱa receptor protein tyrosine phosphatases: mechanistic insights from structural analyses[J]. Semin Cell Dev Biol, 2015, 37: 98–107. DOI:10.1016/j.semcdb.2014.09.007 |
| [4] | Pao LI, Badour K, Siminovitch KA, et al. Nonreceptor protein-tyrosine phosphatases in immune cell signaling[J]. Annu Rev Immunol, 2007, 25: 473–523. DOI:10.1146/annurev.immunol.23.021704.115647 |
| [5] | Hermiston ML, Xu Z, Weiss A. CD45: a critical regulator of signaling thresholds in immune cells[J]. Annu Rev Immunol, 2003, 21: 107–137. DOI:10.1146/annurev.immunol.21.120601.140946 |
| [6] | Vang T, Congia M, Macis MD, et al. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant[J]. Nat Genet, 2005, 37: 1317–1319. DOI:10.1038/ng1673 |
| [7] | Vang T, Landskron J, Viken MK, et al. The autoimmune-predisposing variant of lymphoid tyrosine phosphatase favors T helper 1 responses[J]. Hum Immunol, 2013, 74: 574–585. DOI:10.1016/j.humimm.2012.12.017 |
| [8] | Du J, Qiao Y, Sun L, et al. Lymphoid-specific tyrosine phosphatase (Lyp): a potential drug target for treatment of autoimmune diseases[J]. Curr Drug Targets, 2014, 15: 335–346. DOI:10.2174/13894501113146660236 |
| [9] | Giza S, Goulas A, Gbandi E, et al. The role of PTPN22 C1858T gene polymorphism in diabetes mellitus type 1: first evaluation in Greek children and adolescents[J]. Biomed Res Int, 2013, 2013: 721604. |
| [10] | He Y, Liu S, Menon A, et al. A potent and selective small-molecule inhibitor for the lymphoid-specific tyrosine phos-phatase (LYP), a target associated with autoimmune diseases[J]. J Med Chem, 2013, 56: 4990–5008. DOI:10.1021/jm400248c |
| [11] | Rieck M, Arechiga A, Onengut-Gumuscu S, et al. Genetic variation in PTPN22 corresponds to altered function of T and B lymphocytes[J]. J Immunol, 2007, 179: 4704–4710. DOI:10.4049/jimmunol.179.7.4704 |
| [12] | Yu X, Sun JP, He Y, et al. Structure, inhibitor, and regulatory mechanism of Lyp, a lymphoid-specific tyrosine phosphatase implicated in autoimmune diseases[J]. Proc Natl Acad Sci U S A, 2007, 104: 19767–19772. DOI:10.1073/pnas.0706233104 |
| [13] | Vang T, Xie Y, Liu WH, et al. Inhibition of lymphoid tyrosine phosphatase by benzofuran salicylic acids[J]. J Med Chem, 2011, 54: 562–571. DOI:10.1021/jm101004d |
| [14] | Xie Y, Liu Y, Gong G, et al. Discovery of a novel submicro-molar inhibitor of the lymphoid specific tyrosine phosphatase[J]. Bioorg Med Chem Lett, 2008, 18: 2840–2844. DOI:10.1016/j.bmcl.2008.03.079 |
| [15] | Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays[J]. J Med Chem, 2010, 53: 2719–2740. DOI:10.1021/jm901137j |
| [16] | Wu S, Bottini M, Rickert RC, et al. In silico screening for PTPN22 inhibitors: active hits from an inactive phosphatase conformation[J]. ChemMedChem, 2009, 4: 440–444. DOI:10.1002/cmdc.v4:3 |
| [17] | Vang T, Liu WH, Delacroix L, et al. LYP inhibits T-cell activation when dissociated from CSK[J]. Nat Chem Biol, 2012, 8: 437–446. DOI:10.1038/nchembio.916 |
| [18] | Hou X, Li R, Li K, et al. Fast identification of novel lymphoid tyrosine phosphatase inhibitors using target-ligand interaction-based virtual screening[J]. J Med Chem, 2014, 57: 9309–9322. DOI:10.1021/jm500692u |
| [19] | Stanford SM, Krishnamurthy D, Falk MD, et al. Discovery of a novel series of inhibitors of lymphoid tyrosine phos-phatase with activity in human T cells[J]. J Med Chem, 2011, 54: 1640–1654. DOI:10.1021/jm101202j |
| [20] | Hou X, Li K, Yu X, et al. Protein flexibility in docking-based virtual screening: discovery of novel lymphoid-specific tyrosine phosphatase inhibitors using multiple crystal structures[J]. J Chem Inf Model, 2015, 55: 1973–1983. DOI:10.1021/acs.jcim.5b00344 |
| [21] | Krishnamurthy D, Karver MR, Fiorillo E, et al. Gold (Ⅰ)-mediated inhibition of protein tyrosine phosphatases: a detailed in vitro and cellular study[J]. J Med Chem, 2008, 51: 4790–4795. DOI:10.1021/jm800101w |
| [22] | Karver MR, Krishnamurthy D, Kulkarni RA, et al. Identifying potent, selective protein tyrosine phosphatase inhibitors from a library of Au (Ⅰ) complexes[J]. J Med Chem, 2009, 52: 6912–6918. DOI:10.1021/jm901220m |
| [23] | Karver MR, Krishnamurthy D, Bottini N, et al. Gold (Ⅰ) phosphine mediated selective inhibition of lymphoid tyrosine phosphatase[J]. J Inorg Biochem, 2010, 104: 268–273. DOI:10.1016/j.jinorgbio.2009.12.012 |
| [24] | Kulkarni RA, Stanford SM, Vellore NA, et al. Thiuram disulfides as pseudo-irreversible inhibitors of lymphoid tyrosine phosphatase[J]. ChemMedChem, 2013, 8: 1561–1568. DOI:10.1002/cmdc.201300215 |
| [25] | Ahmed VF, Bottini N, Barrios AM. Covalent inhibition of the lymphoid tyrosine phosphatase[J]. ChemMedChem, 2014, 9: 296–299. DOI:10.1002/cmdc.v9.2 |
| [26] | Zhang XY, Bishop AC. Site-specific incorporation of allosteric-inhibition sites in a protein tyrosine phosphatase[J]. J Am Chem Soc, 2007, 129: 3812–3813. DOI:10.1021/ja069098t |
| [27] | Wiesmann C, Barr KJ, Kung J, et al. Allosteric inhibition of protein tyrosine phosphatase 1B[J]. Nat Struct Mol Biol, 2004, 11: 730–737. DOI:10.1038/nsmb803 |