 2017, Vol. 52
2017, Vol. 52



血管内皮生长因子受体 (vascular endothelial growth factor receptor, VEGFR) 属于蛋白酪氨酸激酶 (protein tyrosine kinases, PTK) 受体超家族[1, 2], 包括VEGFR-1 (Flt-1)、VEGFR-2 (Flk-1/KDR) 和VEGFR-3 (Flt-4)。其中VEGFR-1是造血细胞发育的关键; VEGFR-2主要在血管内皮细胞和造血干细胞中表达; VEGFR-3促进淋巴管内皮细胞的生长。VEGFR-2介导VEGF调控的细胞信号转导通路, 进而刺激血管内皮细胞增殖, 导致血管生长。VEGFR-2在多种恶性肿瘤中高度表达, 包括卵巢癌和甲状腺癌、黑色素瘤和髓母细胞瘤[3]。以VEGFR-2为靶点, 寻找VEGFR-2抑制剂对发现新的抗癌药物提供了良好的途径。
1 VEGFR-2抑制剂的作用机制细胞外配体VEGF与VEGFR-2结合, 诱导VEGFR-2的同源二聚复合物形成, 使其胞内域构象改变, 这些构象的改变使VEGFR-2的酪氨酸残基发生自身磷酸化并将信号传导至下游。其中下游的信号通路包括: Ras (retrovirus associated DNA sequences)/Raf (rapidly accelerated fibrosarcoma)/MAPK (mitogen activated protein kinase) 和PI3K (phosphatidyl inosi tol-3 kinase)/Akt (protein kinase B) 通路[4]。Ras/Raf/MAPK通路被激活后, 最终会激活转录因子 (如Elk1、Etsl、c-Myc等), 从而干扰细胞周期和细胞转化过程, 最终导致肿瘤形成。MAPKs还能诱导蛋白及基质降解、促进细胞迁移、维持肿瘤生长[5, 6]。PI3K/Akt通路被激活后, 通过磷酸化调控多种转录因子 (如FKHRL1、NF-κB、Bcl-2等), 从而抑制凋亡基因的表达。Akt还能磷酸化糖原合成激酶-3 (glycogen synthase kinase3, GSK-3) 和哺乳动物雷帕霉素靶蛋白 (mammalian target of rapamycin, mTOR), 从而上调周期蛋白D (cyclin D), 以及磷酸化一系列抑制蛋白 (如p21CIP1和p27KIP1), 引起细胞周期变短, 从而导致肿瘤发生[7] (图 1)。VEGFR-2抑制剂与ATP竞争而占据其催化域中的ATP结合口袋[8, 9], 抑制VEGF与VEGFR-2的结合而阻断下游信号转导通路。

|
Figure 1 The dimerization of VEGFR-2 and its signal trans-duction pathway |
VEGFR-1、-2、-3具有相似的结构使得VEGFR-2抑制剂可抑制多种VEGFR。然而抑制VEGFR-1、-2能同时抑制肿瘤转移, 且抑制VEGFR-3也能减少肿瘤向淋巴扩散, 这些使得VEGFR-2抑制剂具有更好地抑制肿瘤转移和肿瘤血管生成的效果[10]。同时一些酪氨酸激酶受体家族成员, 包括PDGFRs (platelet derived growth factor receptors)、CSF1R (colony stimulating factor 1 receptor)、c-Kit (stem cell growth factor receptor, SCFR)、FGFR1 (fibroblast growth factor receptor) 和FLT3与VEGFR-2在催化区域具有序列同源性, 因此VEGFR-2抑制剂一般为多靶点抑制剂。其可同时阻断几条对肿瘤转移和肿瘤血管生成重要的独立信号通路。在癌症治疗中, 具有多靶标结合特点的VEGFR-2抑制剂往往比高选择性抑制剂效果更好。
2 VEGFR-2抑制剂的分类VEGFR-2的催化域在N端与C末端之间有一狭长的空腔, 由前后两部分组成。前半部分为ATP的结合位点, 后半部分则包含自磷酸化区域[11]。VEGFR-2抑制剂与VEGFR-2的主要结合区域包括疏水腔Ⅰ (hydrophobic region Ⅰ) (周边的疏水氨基酸是Leu-840、Phe-918和Gly-922), 疏水腔Ⅱ (hydrophobic region Ⅱ) (周边的疏水氨基酸是Leu-889、Ile-892、Val-898和Ile-1044) 和中间的链接处 (linker region) (周边的疏水氨基酸是Ala-866、Val-914、Leu-1035和Cys-1045)[12-14] (图 2)。其中疏水腔后有两个外部亲水区域, 分别是外部亲水腔Ⅰ (extra hydrophilic region Ⅰ) (靠近疏水腔Ⅰ) 和外部亲水腔Ⅱ (extra hydrophilic region Ⅱ) (靠近疏水腔Ⅱ)。

|
Figure 2 The binding regions between VEGFR-2 inhibitor and VEGFR-2 |
根据小分子抑制剂与VEGFR-2结合方式, 通常将这些小分子分为以下几类: Ⅰ类激酶抑制剂、新型Ⅰ类激酶抑制剂、Ⅱ类激酶抑制剂和新型Ⅱ类激酶抑制剂。Ⅰ类激酶抑制剂与ATP是竞争性关系, 其多通过杂环占据疏水腔Ⅰ而与氨基酸残基Glu-917和Glu-919形成氢键以及与疏水腔Ⅰ发生疏水作用[15] (图 3A)。新型Ⅰ类激酶抑制剂能与氨基酸残基Cys-919形成氢键, 此外在Ⅰ类激酶抑制剂结构的基础上多了一个化学结构片段, 能与口袋狭长空腔的氨基酸残基Asp-1046和Glu-885有氢键作用 (图 3B), 但其不能够有效占据疏水腔Ⅱ[16]。Ⅱ类激酶抑制剂与ATP是非直接竞争性的关系, 因为其骨架和激酶的结合区域 (疏水区Ⅰ, 铰链和疏水区Ⅱ) 与ATP和激酶的结合区域 (疏水区Ⅰ, 即ATP结合区中的腺嘌呤区) 只是部分重合[17]。Ⅱ类激酶抑制剂除了能与氨基酸残基Cys-919形成氢键从而占据疏水腔Ⅰ外, 还占据疏水腔Ⅱ。Ⅱ类激酶抑制剂的特点是通常由脲、酰胺或者其电子等排体作为桥梁来连接占据疏水腔Ⅰ和疏水腔Ⅱ的两个芳环。且该化学连接片段与氨基酸残基Asp-1046和Glu-885有氢键作用 (以sorafenib为例, 其与VEGFR-2的激酶口袋作用) (图 3C)。另外由于Ⅱ类激酶抑制剂能够占据结构相对不太保守的疏水腔Ⅱ, 因此提高了选择性[16]。新型Ⅱ类激酶抑制剂则是在Ⅱ类激酶抑制剂原有占据疏水腔Ⅱ的芳环上引入一个结构片段来占据疏水腔Ⅱ外的亲水腔Ⅱ (图 3D)。尽管这个化学结构片段可与此亲水腔存在弱的离子和极性相互作用, 但对整个分子与靶点结合力的贡献较小[18, 19]。通过此片段的修饰, 能够改善小分子的透膜性及选择性[20, 21]。根据以上对小分子抑制剂的分类, 本文将分别叙述近些年来较有代表性的VEGFR-2小分子抑制剂。

|
Figure 3 The crystal structures of VEGFR-2 complexed with various VEGFR-2 inhibitors |
化合物1 (sunitinib, 表 1) 由Pfizer公司开发, 其对VEGFR-2的IC50为9 nmol·L-1, 是典型的Ⅰ类激酶抑制剂, 2006年由FDA批准治疗胃肠癌和晚期肾癌, 能延长腺泡状软组织肉瘤患者19个月的时间。化合物2 (vandetanib) 由AstraZeneca开发, 对VEGFR-2的IC50为40 nmol·L-1, 在2011年由FDA批准用于治疗晚期不适合手术的骨髓癌与甲状腺癌[22]。化合物3 (anlotinib) 是一种口服的VEGFR抑制剂, 由江苏正大天晴开发, 在临床Ⅱ/Ⅲ期进行转移性结直肠癌的试验。临床Ⅱ试验治疗晚期非小细胞肺癌 (NSCLC)、晚期软组织肉瘤、晚期甲状腺髓样癌和晚期肾细胞癌[23]。试验表明, anlotinib能够平均延长甲状腺癌患者12.8个月的时间。化合物4 (cediranib) 由AstraZeneca公司开发, 其对VEGFR-2的IC50为4 nmol·L-1。2010年3月8日, AstraZeneca宣布cediranib在结直肠癌临床Ⅲ期失败。化合物5 (nintedanib) 由Boehringer Ingelheim Pharma GmbH & Co.公司开发, 是针对VEGFR、PDGFR和FGFR的多靶点抑制剂。其对VEGFR-1、-2及-3的IC50分别是104、5及5 nmol·L-1[24]。试验结果表明其能延长原发性腹膜癌患者6个月的时间。FDA于2014年10月15日批准了nintedanib对于特发性肺纤维化 (idiopathic pulmonary fibrosis) 的治疗[25]。化合物6 (semaxinib) 由Sugen公司开发, 对VEGFR-2的IC50为10 nmol·L-1。Semaxinib对VEGF所依赖的人类脐静脉内皮细胞具有非常高的选择性, 它对此类细胞的IC50为40 nmol·L-1, 而对FGF所依赖的人类脐静脉内皮细胞的IC50为50 μmol·L-1, 二者相差了1 000多倍。化合物7由Bristol-Myers Squibb开发, 此化合物对VEGFR-1、VEGFR-2和FGFR-1都有一定的抑制作用。对无胸腺老鼠移植的人类肺癌模型H3396和L2987具有非常好的体内活性。但此化合物在药代动力学和理化性质方面表现不足, 例如其水溶性差。为克服此困难研发了其前药BMS-582664 (brivanib alaninate, 化合物8)[26], 为化合物7的二级醇与可代谢的酯基相连接。Brivanib alaninate具有非常好的理化性, 同时具有显著的抗癌活性, 尤其针对无胸腺老鼠移植的人类肺癌模型L2987[26, 27]。
| Table 1 Type Ⅰ inhibitors |
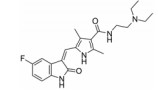
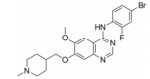
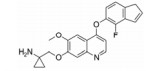
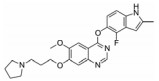
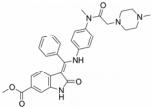
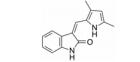
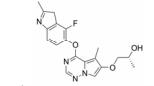
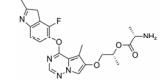
化合物9 (lenvatinib, 商品名: Lenvima, 表 2) 由美国FDA于2015年批准其治疗侵袭性、分化型甲状腺癌 (DTC) 患者, 适用于在接受放射性碘治疗后疾病仍恶化 (放射性碘难治性疾病) 的患者。Lenvatinib是一种多靶点抑制剂, 作用于VEGFR-2、-3的IC50分别为4、5.2 nmol·L-1, 作用于VEGFR-2、-3比作用于FGFR1和PDGFRα/β选择性高10倍左右。Lenvatinib有效抑制血管生成, 也显著抑制VEGF/KDR和SCF/Kit信号通路。Lenvatinib分别作用于由VEGF和VEGF-C刺激的HUVECs, 能有效抑制VEGFR-2和VEGFR-3磷酸化, IC50分别为0.83和0.36 nmol·L-1[28]。最新研究显示用1 μmol·L-1的Lenvatinib通过抑制FGFR和PDGFR信号通路, 从而明显抑制细胞迁移和入侵[29]。化合物10 (lucitanib) 是由中国科学院上海药物研究所等机构研发, 针对的适应证是小细胞肺癌、非小细胞肺癌、乳腺癌等, 其同时也是多靶点抑制剂, 对3种形式的VEGFR (VEGFR-1、-2、-3)、FGFR2、CSF1R (c-FMS)、FGFR1等都有抑制效果。Lucitanib正在进行Ⅰ/Ⅱ期临床研究中。Lucitanib对静脉内皮细胞的IC50为1.00 nmol·L-1。在酶水平上, 对VEGFR-1 (Flt-1)、VEGFR-3 (FLT4)、FGFR1的IC50分别为0.162 μmol·L-1、34、58 nmol·L-1, 对VEGFR-2 (FLK-1/KDR) 的IC50 < 30 nmol·L-1[30-32]。化合物11 (fruquintinib) 是一种新型小分子化合物, 其能选择性抑制VEGFR。目前它处在临床研究Ⅲ期, 用于治疗局部晚期或转移性结直肠癌, Ⅱ期临床试验也进行了非小细胞肺癌的治疗。Fruquintinib的作用机制是同时抑制VEGFR-1、-2、-3。其疗效显著, 具有广谱抗肿瘤活性。化合物12 (pazopanib) 由GSK公司开发, 其对VEGFR-2的IC50为30 nmol·L-1, 在2009年被FDA批准用于治疗晚期肾癌[33]。化合物13 (axitinib, 阿西替尼) 由Pfizer开发研制, 商品名为Inlyta。Axitinib于2012年1月27日被FDA批准上市, 用于其他手段治疗无效的晚期肾癌 (renal cell carcinoma, RCC)。Axitinib与Pfizer的另一抗癌药物舒尼替尼 (sunitinib) 类似, Axitinib也是多靶点酪氨酸激酶抑制剂, 可以抑制3种形式的VEGFR (VEGFR-1、-2、-3)、PDGFR和c-Kit[34]。目前该药针对的适应证为转移性乳腺癌、胰腺内分泌肿瘤、多形性胶质母细胞瘤、前列腺癌、黑色素瘤、甲状腺软组织肉瘤、非小细胞肺癌 (NSCLC)、头部和颈部癌症、遗传性疾病、神经系统、肾 (肾细胞癌)、胃肿瘤、家族性嗜铬细胞瘤、副神经节瘤、大肠癌、间皮瘤[35]。
| Table 2 Novel type Ⅰ inhibitors |
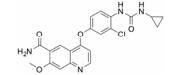
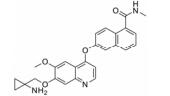
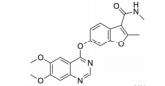
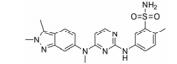
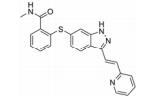
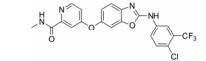
化合物14 (sorafenib, 表 3) 由Bayer公司开发, 其对VEGFR-2的IC50为90 nmol·L-1。Sorafenib是典型的Ⅱ类激酶抑制剂, 由FDA批准用于治疗晚期肾癌[36]。通过将sunitinib (化合物1) 与sorafenib进行对比, 表明Ⅱ类激酶抑制剂的选择性优于Ⅰ类激酶抑制剂。Sunitinib作为Ⅰ类激酶抑制剂能够与113种激酶中的73种相结合, 而sorafenib作为Ⅱ类激酶抑制剂却只与113种激酶中的35种相结合[37]。化合物15 (regorafenib, Stivarga®) 是由Bayer和美国生技制药公司联合开发, 一种新型的多激酶抑制剂, 通过抑制多种促进肿瘤生长蛋白质激酶, 靶向作用于肿瘤生成、肿瘤血管发生[38]。它对VEGFR-1、VEGFR-3、PDGFRβ、FGFR1和Tie-2以及突变的致癌基因激酶Kit、RET和B-Raf的IC50都处于非常低的纳摩尔浓度水平[39]。2012年9月27日, FDA批准了口服药物regorafenib用于治疗既往接受过或以氟尿嘧啶、奥沙利铂和伊立替康为基础的化疗、抗VEGF治疗, 以及抗EGFR治疗转移性结直肠癌 (CRC) 患者。2013年2月28日FDA批准regorafenib片剂用于治疗先前接受过伊马替尼和舒尼替尼治疗的局部晚期患者, 以及不能手术切除或转移性胃肠道间质瘤 (GIST) 患者。临床前研究已经证明regorafenib能够抑制在肿瘤新生血管发生 (新血管生长) 中起重要作用的几种促血管生成的VEGF受体激酶。它还抑制几种致癌和肿瘤微环境激酶, 包括Kit和PDGFR, 在GIST的发生和复发中起关键作用。Regorafenib用于服用伊马替尼 (格列卫) 和舒尼替尼 (索坦) 无效的胃肠道间质瘤患者的效果要强于转移性结直肠癌患者。化合物16 (cabozantinib, XL184, BMS-907351) 由Exelixis开发。其针对的靶点为VEGFR-2、MET和RET[40]。此抑制剂对Kit、FLT3和Tek的IC50的活性值处于非常低的纳摩尔浓度级别。其针对的适应证为甲状腺髓样癌、胶质母细胞瘤、甲状腺髓样癌和前列腺癌[41, 42]。Cabozantinib是一种口服药物, 通过靶向抑制MET、VEGFR2及RET信号通路而发挥抗肿瘤作用, 它能够杀死肿瘤细胞, 减少转移并抑制血管生成[43]。Cabozantinib于2013年以孤儿药的身份获得FDA的批准用于治疗甲状腺髓样癌。段文虎课题组报道了一系列由Cabozantinib改造而来的化合物[44]。这些化合物同时抑制c-Met/VEGFR-2双靶点。化合物17 (tivozanib, KRN951、AV-951) 由Astellas开发。此化合物中脲结构与激酶催化位点有氢键作用。体外激酶选择性活性筛选实验表明, tivozanib对VEGFR-1、VEGFR-2、VGFR-3等同类别酪氨酸激酶的磷酸化具有显著抑制作用, IC50值分别为30、6.5和15 nmol·L-1。同时对EphB2、PDGFR-α、PDGFR-β、c-Kit和Tie2等其他酪氨酸激酶的磷酸化也具有显著抑制作用, IC50值分别为24、40、49、78和78 nmol·L-1。体外细胞激酶选择性活性筛选实验显示, tivozanib显著抑制由VEGFR-1、VEGFR-2、VEGFR-3、c-Kit和PDGFR-β刺激的人脐静脉血管内皮细胞 (HUVEC) 的磷酸化, IC50值分别为0.21、0.16、0.24、1.63和1.72 nmol·L-1, 而对FGFR-1、FLT3、EGFR、IGF-1和C-Met磷酸化的抑制作用较弱[45]。此外, 在VEGFR刺激的HUVEC迁移实验中, 1 nmol·L-1 tivozanib能抑制约40%的HUVEC迁移, 大于10 nmol·L-1时, 完全抑制HUVEC迁移; 在细胞毒性实验中, tivozanib给药浓度低于1 μmol·L-1时, 对多种癌细胞均没有明显的细胞毒性, 表明它的主要作用是抑制血管生成[45]。它对肺癌、乳腺癌、结肠癌、卵巢癌、前列腺癌和胰腺癌都有一定的抗癌活性。临床Ⅱ期表明, tivozanib对晚期肾癌的患者有很好的效果。Ⅲ期临床证明tivozanib较sorafenib能够平均延长晚期肾癌患者3个月的时间。化合物18 (foretinib) 由Exelixis开发, 在2007年由GSK注册。其特点是7-甲氧基上的吗啉基团作为亲水性侧链。它是多靶点抑制剂, 对VEGFR和MET有显著的抑制效果, 对VEGFR-1、VEGFR-2、VEGFR-3的IC50值分别为0.8、6.8、2.8 nmol·L-1[46], 对MET的IC50为0.5 nmol·L-1。它同时对PDGFRs和促血管新生蛋白因子受体Tie-2有一定的抑制作用, 对FGFR和EGFR也有作用。在人的脐静脉血管内皮细胞 (HUVEC) 上的IC50为16 nmol·L-1[46]。Ⅱ期临床表明foretinib对于晚期乳头状肾细胞癌患者具有活性, 并且其毒性可控, 对存在胚系MET基因突变的患者具有较高的缓解率。也有研究报道通过修饰占据疏水腔Ⅱ的化学片段来改变小分子对激酶的选择性。化合物19 (telatinib, BAY 57-9352) 由Bayer公司研发。它的核心骨架是五元并邻双氮哒嗪[38]。它对VEGFR、PDGFR和c-Kit都有抑制作用。其对VEGFR-1和VEGFR-2的IC50分别是6和4 nmol·L-1[38], 它对人类静脉内皮细胞有抑制作用, 尽管它对肿瘤细胞没有直接的抑制。药代动力学研究表示在临床Ⅱ期, telatinib推荐的使用剂量是每天2次, 一次900 mg。临床Ⅱ期的研究表明其可作为一线治疗药物联合其他化学疗法用在晚期胃癌的治疗中[47]。化合物20 (vatalanib) 对VEGFR-1、-2、-3的IC50值分别是110、43、195 nmol·L-1[24]。对PDGFRβ和c-Kit也有一定的抑制。分子对接研究表明, 对氯苯胺基团在疏水腔Ⅱ中[24], 苯胺的NH基团通过水分子介导与铰链上的Glu915和Cys917有氢键作用, 吡啶上的氮与Lys1060有氢键作用。其对恶性血液病的治疗处在临床Ⅰ、Ⅱ期的研究阶段, 同时针对晚期结肠癌的治疗处在临床Ⅲ期的研究阶段[48]。研究表明vatalanib是治疗结肠癌的一线药物。虽然Ⅱ类激酶抑制剂选择性高, 然而其抗耐药性则没有Ⅰ类激酶抑制剂好。这是因为当激酶在ATP结合区域发生突变时, 尽管会对Ⅰ类激酶抑制剂结合变弱, 但也可能使得其与ATP结合也同样变弱, 致使激酶突变体的催化活性降低。而当非ATP的结合区域发生突变, 如果恰好发生在与Ⅱ类激酶抑制剂的结合区域, 这将导致激酶仍保持其与ATP结合后的催化活性, 但却对Ⅱ类激酶抑制剂产生了耐药性[12]。
| Table 3 Type Ⅱ inhibitors |
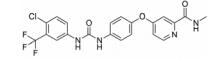
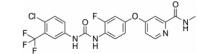
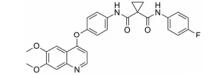

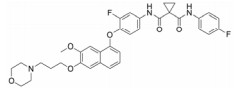
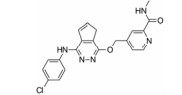
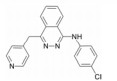
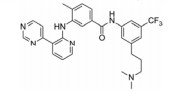
化合物21 (ponatinib, 表 4) 于2013年上市, 用于治疗淋巴细胞性白血病, 主要针对c-Abl靶点[49]。近来也发现其对VEGFR-2的IC50为1.5 nmol·L-1有很好的活性。化合物22由Amgen公司开发, 对VEGFR-2的IC50为8.7 nmol·L-1, 而其对HUVEC的IC50 > 1 140 nmol·L-1[50]。这可能与化合物的透膜性有关, 当将取代的三氟甲基替换为含氮片段的化合物23, 改善了化合物的亲水性, 化合物23在酶上的活性尽管没有什么改变, 但在细胞上的活性却提高了76倍[50]。化合物24由Amgen公司开发, 对VEGFR-2的IC50为2 nmol·L-1[51]。
| Table 4 Novel type Ⅱ inhibitors |
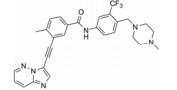

VEGFR已经成为抑制肿瘤生长的有效靶点, 近年来针对VEGFR的抑制剂仍然不断涌现。仅自2009年以来, 就有4个VEGFR抑制剂获批而成功上市, 表明这些抑制剂有很高的治疗价值, 且在未来癌症治疗领域具有极其重要的研究意义。这些抑制剂的共同特点是, 它们不只仅仅作用于VEGFR, 而往往是作用于多个靶点, 这给研发抗肿瘤药物提供了许多启示。由于癌症的复杂性, 涉及到多个信号通路。因此, 有选择性的多靶点抑制剂通过抑制肿瘤发生发展过程中多个重要激酶的活性往往能够取得更好的癌症治疗效果。然而针对VEGFR-2激酶抑制剂的弱选择性却也有可能导致不良反应。如服用索拉菲尼会导致皮肤毒性、消化道反应、肝功能损伤、血管系统毒性反应和全身反应等[52]。因此所谓的多靶点抑制并不是一味地抑制多种激酶, 而应该是针对癌症的特点选择性地抑制某些激酶。
从临床和生物化学的角度来看, 还有许多尚未被完全探索的问题。关于临床方面, 已有文献报道考虑到使用这些激酶抑制剂的疾病的严重性, 认为VEGFR抑制剂的毒性特征通常是可接受和可控的。然而, 其中部分不良反应却是比较严重或危及生命的, 需要停止治疗。VEGFR抑制剂正与常规化疗进行组合使用, 往往能延长患者存活时间并减少化疗的剂量。但在采取联合用药策略时需要予以特别重视以避免产生严重影响。另外, 许多证据表明单独使用VEGFR抑制剂通常不足以阻断和根除肿瘤, 而本文提到的大部分活性化合物由于其多靶点结合的特点, 能抑制涉及血管生成和细胞生长的多个激酶, 就具有比较好的治疗效果。但仍需要深入研究这些激酶的同源性与差异性, 并同时考虑特定肿瘤自身发生和发展特点, 而设计具有选择性的多激酶抑制剂。随着新技术的发展, 尤其是化学、计算机和生物学科的发展, 对系统地研究疾病的发病机制、生物通路、激酶的差异性提供了更多的帮助, 这些也将有助于研发出更有效的新型VEGFR抑制剂。
| [1] | Risau W. Mechanisms of angiogenesis[J]. Nature, 1997, 386: 671–674. DOI:10.1038/386671a0 |
| [2] | Folkman J, D'Amore PA. Blood vessel formation:what is its molecular basis?[J]. Cell, 1996, 87: 1153–1155. DOI:10.1016/S0092-8674(00)81810-3 |
| [3] | Cristina RA, Pallares J, Cristina MC, et al. Overexpression and activation of EGFR and VEGFR2 in medullary thyroid carcinomas is related to metastasis[J]. Endocr Relat Cancer, 2010, 17: 7–16. DOI:10.1677/ERC-08-0304 |
| [4] | Holmes K, Roberts OL, Thomas AM, et al. Vascular endothelial growth factor receptor-2:structure, function, intracellular signalling and therapeutic inhibition[J]. Cell Signal, 2007, 19: 2003–2012. DOI:10.1016/j.cellsig.2007.05.013 |
| [5] | Liebmann C. Regulation of MAP kinase activity by peptide receptor signalling pathway:paradigms of multiplicity[J]. Cell Signal, 2001, 13: 777–785. DOI:10.1016/S0898-6568(01)00192-9 |
| [6] | Mao YJ, Li HH, Li JF, et al. Signal transduction by protein tyrosine kinases and antitumor agents[J]. Acta Pharm Sin (药学学报), 2008, 43: 323–334. |
| [7] | Shaw RJ, Cantley LC. Ras, PI (3) K and mTOR signalling controls tumour cell growth[J]. Nature, 2006, 441: 424–430. DOI:10.1038/nature04869 |
| [8] | Boyer SJ. Small molecule inhibitors of KDR (VEGFR-2) kinase:an overview of structure activity relationships[J]. Curr Top Med Chem, 2002, 2: 973–1000. DOI:10.2174/1568026023393273 |
| [9] | Schenone S, Bondavalli F, Botta M. Antiangiogenic agents:an update on small molecule VEGFR inhibitors[J]. Curr Med Chem, 2007, 14: 2495–2516. DOI:10.2174/092986707782023622 |
| [10] | Gille J, Heidenreich R, Pinter A, et al. Simultaneous blockade of VEGFR-1 and VEGFR-2 activation is necessary to efficiently inhibit experimental melanoma growth and metastasis formation[J]. Int J Cancer, 2007, 120: 1899–1908. DOI:10.1002/(ISSN)1097-0215 |
| [11] | Huse M, Kuriyan J. The conformational plasticity of protein kinases[J]. Cell, 2002, 109: 275–282. DOI:10.1016/S0092-8674(02)00741-9 |
| [12] | Zuccotto F, Ardini E, Casale E, et al. Through the "gatekeeper door":exploiting the active kinase conformation[J]. J Med Chem, 2010, 53: 2681–2694. DOI:10.1021/jm901443h |
| [13] | Backes AC, Zech B, Felber B, et al. Small-molecule inhibitors binding to protein kinase. Part Ⅱ:the novel pharmacophore approach of type Ⅱ and type Ⅲ inhibition[J]. Expert Opin Drug Discov, 2008, 3: 1427–1449. DOI:10.1517/17460440802580106 |
| [14] | Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations[J]. Nat Chem Biol, 2006, 2: 358–364. DOI:10.1038/nchembio799 |
| [15] | Gray NS, Wodicka L, Thunnissen A-MWH, et al. Exploiting chemical libraries, structure, and genomics in the search for kinase inhibitors[J]. Science, 1998, 281: 533–538. DOI:10.1126/science.281.5376.533 |
| [16] | Mol CD, Fabbro D, Hosfield DJ. Structural insights into the conformational selectivity of STI-571 and related kinase inhibitors[J]. Curr Opin Drug Discov Devel, 2004, 7: 639–648. |
| [17] | Pargellis C, Tong L, Churchill L, et al. Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site[J]. Nat Struct Biol, 2002, 9: 268–272. DOI:10.1038/nsb770 |
| [18] | Keri G, Orfi L, Eros D, et al. Signal transduction therapy with rationally designed kinase inhibitors[J]. Curr Signal Transduction Ther, 2006, 1: 67–95. DOI:10.2174/157436206775269190 |
| [19] | Fabbro D, Ruetz S, Buchdunger E, et al. Protein kinases as targets for anticancer agents:from inhibitors to useful drugs[J]. Pharmacol Ther, 2002, 93: 79–98. DOI:10.1016/S0163-7258(02)00179-1 |
| [20] | Kontopidis G, McInnes C, Pandalaneni SR, et al. Differential binding of inhibitors to active and inactive CDK2 provides insights for drug design[J]. Chem Biol, 2006, 13: 201–211. DOI:10.1016/j.chembiol.2005.11.011 |
| [21] | Wang Z, Canagarajah BJ, Boehm JC, et al. Structural basis of inhibitor selectivity in MAP kinases[J]. Structure, 1998, 6: 1117–1128. DOI:10.1016/S0969-2126(98)00113-0 |
| [22] | Wedge SR, Ogilvie DJ, Dukes M, et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration[J]. Cancer Res, 2002, 62: 4645–4655. |
| [23] | Sun YK, Niu W, Chi Y, et al. Safety, pharmacokinetic, and antitumor activity of anlotinib, an oral multi-target tyrosine kinase inhibitor, in patients with advanced refractory solid tumors (Annual Meeting of the American-Society-of-Clinical-Oncology)[C]. Chicago, IL:2015:33. |
| [24] | Bold G, Altmann KH, Frei J, et al. New anilinophthalazines as potent and orally well absorbed inhibitors of the VEGF receptor tyrosine kinases useful as antagonists of tumor-driven angiogenesis[J]. J Med Chem, 2000, 43: 2310–2323. DOI:10.1021/jm9909443 |
| [25] | Ledermann JA, Hackshaw A, Kaye S, et al. Randomized phase Ⅱ placebo-controlled trial of maintenance therapy using the oral triple angiokinase inhibitor BIBF 1120 after chemotherapy for relapsed ovarian cancer[J]. J Clin Oncol, 2011, 29: 3798–3804. DOI:10.1200/JCO.2010.33.5208 |
| [26] | Gong J, Gan J, Masson E, et al. Metabolic chiral inversion of brivanib and its relevance to safety and pharmacology[J]. Drug Metab Dispos, 2012, 40: 2374–2380. DOI:10.1124/dmd.112.047340 |
| [27] | Mekhail T, Masson E, Fischer BS, et al. Metabolism, excretion, and pharmacokinetics of oral brivanib in patients with advanced or metastatic solid tumors[J]. Drug Metab Dispos, 2010, 38: 1962–1966. DOI:10.1124/dmd.110.033951 |
| [28] | Gild ML, Bullock M, Robinson BG, et al. Multikinase inhibitors:a new option for the treatment of thyroid cancer[J]. Nat Rev Endocrinol, 2011, 7: 617–624. DOI:10.1038/nrendo.2011.141 |
| [29] | Yamada K, Yamamoto N, Yamada Y, et al. Phase Ⅰ dose-escalation study and biomarker analysis of E7080 in patients with advanced solid tumors[J]. Clin Cancer Res, 2011, 17: 2528–2537. DOI:10.1158/1078-0432.CCR-10-2638 |
| [30] | Guffanti F, Chila R, Bello E, et al. Preclinical activity of the VEGFR, FGFR and PDGFR inhibitor lucitanib in FGFR2 aberrant endometrial and gastric cancer models[J]. Eur J Cancer, 2014, 50: 110–116. |
| [31] | Holmstoem T, Moilanen A, Linnanen T, et al. ODM-203, a novel, selective and balanced FGFR and VEGFR inhibitor with strong anti-tumor activity in FGFR-and VEGFR-dependent cancer models[J]. Eur J Cancer, 2014, 50: 142. |
| [32] | Nguyen M, Lin KK, Burbridge MF, et al. Nonclinical activity of the FGFR, VEGFR and PDGFR inhibitor lucitanib in FGFR3 translocated tumor models (106th Annual Meeting of the American-Association-for-Cancer-Research)[C]. Philadelphia, PA:Cancer Research, 2015:75. |
| [33] | Podar K, Tonon G, Sattler M, et al. The small-molecule VEGF receptor inhibitor pazopanib (GW786034B) targets both tumor and endothelial cells in multiple myeloma[J]. Proc Natl Acad Sci U S A, 2006, 103: 19478–19483. DOI:10.1073/pnas.0609329103 |
| [34] | Ho TH, Jonasch E. Axitinib in the treatment of metastatic renal cell carcinoma[J]. Future Oncol, 2011, 7: 1247–1253. DOI:10.2217/fon.11.107 |
| [35] | Solowiej J, Bergqvist S, McTigue MA, et al. Characterizing the effects of the juxtamembrane domain on vascular endothelial growth factor receptor-2 enzymatic activity, autophosphorylation, and inhibition by axitinib[J]. Biochemistry, 2009, 48: 7019–7031. DOI:10.1021/bi900522y |
| [36] | Levine AM, Tulpule A, Quinn DI, et al. Phase Ⅰ study of antisense oligonucleotide against vascular endothelial growth factor:decrease in plasma vascular endothelial growth factor with potential clinical efficacy[J]. J Clin Oncol, 2006, 24: 1712–1719. DOI:10.1200/JCO.2005.03.4801 |
| [37] | Fabian MA, Biggs WH, Treiber DK, et al. A small molecule-kinase interaction map for clinical kinase inhibitors[J]. Nat Biotechnol, 2005, 23: 329–336. DOI:10.1038/nbt1068 |
| [38] | Wilhelm S, Dumas J, Ladouceur G, et al. Preparation of diaryl ureas with kinase inhibiting activity:US, WO2004113274A2[P]. 2004-05-19. |
| [39] | Hwang SH, Wecksler AT, Zhang G, et al. Synthesis and biological evaluation of sorafenib-and regorafenib-like sEH inhibitors[J]. Bioorg Med Chem Lett, 2013, 23: 3732–3737. DOI:10.1016/j.bmcl.2013.05.011 |
| [40] | Yakes FM, Chen J, Tan J, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth[J]. Mol Cancer Ther, 2011, 10: 2298–2308. DOI:10.1158/1535-7163.MCT-11-0264 |
| [41] | Zhang Y, Guessous F, Kofman A, et al. XL-184, a MET, VEGFR-2 and RET kinase inhibitor for the treatment of thyroid cancer, glioblastoma multiforme and NSCLC[J]. IDrugs, 2010, 13: 112–121. |
| [42] | Bowles DW, Kessler ER, Jimeno A. Multi-targeted tyrosine kinase inhibitors in clinical development:focus on XL-184(cabozantinib)[J]. Drugs Today, 2011, 47: 857–868. DOI:10.1358/dot.2011.47.11.1688487 |
| [43] | Durante C, Russo D, Verrienti A, et al. XL184(cabozantinib) for medullary thyroid carcinoma[J]. Expert Opin Invest Drugs, 2011, 20: 407–413. DOI:10.1517/13543784.2011.559163 |
| [44] | Zhan Z, Ai J, Liu Q, et al. Discovery of anilinopyrimidines as dual inhibitors of c-Met and VEGFR-2:synthesis, SAR, and cellular activity[J]. ACS Med Chem Lett, 2014, 5: 673–678. DOI:10.1021/ml500066m |
| [45] | Dumas J. Protein kinase inhibitors from the urea class[J]. Curr Opin Drug Discov Devel, 2002, 5: 718–727. |
| [46] | Huynh H, Ong R, Soo KC. Foretinib demonstrates anti-tumor activity and improves overall survival in preclinical models of hepatocellular carcinoma[J]. Angiogenesis, 2012, 15: 59–70. DOI:10.1007/s10456-011-9243-z |
| [47] | Eskens FALM, Steeghs N, Verweij J, et al. Phase Ⅰ dose escalation study of telatinib, a tyrosine kinase inhibitor of vascular endothelial growth factor receptor 2 and 3, platelet-derived growth factor receptor β, and c-Kit, in patients with advanced or metastatic solid tumors[J]. J Clin Oncol, 2009, 27: 4169–4176. DOI:10.1200/JCO.2008.18.8193 |
| [48] | Joensuu H, De BF, Grignagni G, et al. Vatalanib for metastatic gastrointestinal stromal tumour (GIST) resistant to imatinib:final results of a phase Ⅱ study[J]. Br J Cancer, 2011, 104: 1686–1690. DOI:10.1038/bjc.2011.151 |
| [49] | Huang WS, Metcalf CA, Sundaramoorthi R, et al. Discovery of 3-[2-(imidazo[1, 2-b]pyridazin-3-yl) ethynyl]-4-methyl-N-{4-[(4-methylpiperazin-1-yl) methyl]-3-(trifluoromethyl) phenyl} benzamide (AP24534), a potent, orally active pan-inhibitor of breakpoint cluster region-abelson (BCR-ABL) kinase including the T315I gatekeeper mutant[J]. J Med Chem, 2010, 53: 4701–4719. DOI:10.1021/jm100395q |
| [50] | Potashman MH, Bready J, Coxon A, et al. Design, synthesis, and evaluation of orally active benzimidazoles and benzoxazoles as vascular endothelial growth factor-2 receptor tyrosine kinase inhibitors[J]. J Med Chem, 2007, 50: 4351–4373. DOI:10.1021/jm070034i |
| [51] | Hodous BL, Geuns-Meyer SD, Hughes PE, et al. Evolution of a highly selective and potent 2-(pyridin-2-yl)-1, 3, 5-triazine tie-2 kinase inhibitor[J]. J Med Chem, 2007, 50: 611–626. DOI:10.1021/jm061107l |
| [52] | Yao JW, Sun W, Chen J, et al. Advances in the study of structural modifications of multi-target anticancer drug sorafenib[J]. Acta Pharm Sin (药学学报), 2012, 47: 1111–1119. |