 2017, Vol. 52
2017, Vol. 52

编者按:
新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
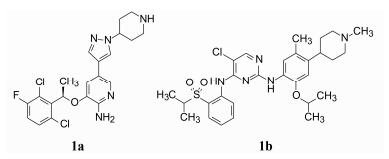
色瑞替尼是首创药物克唑替尼的后续药, 作用机制明确, 靶标相同, 二者上市相距三年。实际上, 色瑞替尼貌似跟踪, 却胜似跟踪, 因为它对克唑替尼发生耐药的患者效果显著而有重大突破, 具有鲜明的创新性。尽管迄今尚不完全清楚其抗耐药性的结构基础, 发明色瑞替尼包含有幸运的色彩, 但诺华的研究者将先导物的结构“精雕细刻”地优化, 为了在酶和细胞水平上提高活性和选择性, 消除警戒结构和强化成药性, 对遍及骨架周遭的基团和片段进行了多方位变换和探索, 成为运用药物化学理念和构效关系分析的一个成功范例。
1 研究背景有一种肿瘤称作间变性大细胞淋巴瘤 (ALCL), 是类罕见的非何杰金T细胞淋巴瘤, 多发生在皮肤、骨骼、软组织和多种脏器中, 这种全身性肿瘤的发生原因, 大多是发生了t (2;5) (p23;q25) 染色体易位, 产生了由间变性淋巴瘤激酶 (anaplastic lymphoma kinase, ALK) 与核磷蛋白 (nucleophosmin, NPM) 胞内结构域的融合基因, 高表达的ALK-NPM蛋白是发生间变性淋巴瘤、炎性肌纤维细胞瘤和非小细胞肺癌等关键性酶, 因而是研发这类罕见病个性化治疗的关键性靶标。
以ALK激酶为靶标首创性的药物是由辉瑞研发的克唑替尼 (1a, crizotinib), 上市于2011年, 治疗非小细胞肺癌。诺华创制的本品色瑞替尼 (1b, ceritinib), 上市于2014年, 三年的间隔使色瑞替尼沦为后续研发的药物, 然而由于与克唑替尼有相当长时间的研发重合期, 他们的研发路径和结构优化过程以及候选化合物的确定, 却是独立进行和较少借鉴的, 反映在结构上, 1a和1b具有不同的结构骨架和药效团的特征。而且, 尽管是作用于同一靶标, 但本品的特点是对克唑替尼发生耐药的患者有效, 被称为抢救性抗肿瘤药物。
|
色瑞替尼毕竟是晚于首创药物克唑替尼的第二代药物, 但由于起始的先导物的活性很高, 起点高, 因而评价优化的化合物活性在用ALK-NPM激酶测定的同时, 还用高表达ALK-NPM激酶的Ba/F3细胞评价抑制生长活性 (目标是酶和细胞水平的高活性); 也由于ALK激酶属于胰岛素受体家族的成员, 为了评价化合物对ALK的特异性抑制, 还评价对高表达Tel-InsR融合蛋白的Ba/F3细胞的作用 (目标是低或无活性); 为了消除细胞毒的非特异性作用, 还测定了对野生型 (WT) Ba/F3细胞的活性 (目标是低或无活性); 还用具有表达ALK-NPM蛋白、人ALKL肿瘤的ALK呈阳性的Karpas 299细胞评价化合物的抗增殖活性 (目标是高活性)。
3 先导化合物的发现 3.1 具有高活性化合物的发现诺华公司为了研发针对ALK-NPM激酶为靶标的抑制剂, 用已有针对其他激酶而合成的小分子化合物库进行了随机筛选, 从中发现了化合物2 (代号TAE684) 对高表达ALK-NPM的Ba/F3细胞有强抑制活性, IC50=3 nmol·L-1, 而对野生型Ba/F3细胞在1 μmol·L-1浓度下没有抑制作用, 提示该先导物是选择性高的强效抑制剂。图 1是化合物2与ALK激酶分子对接图。

|
图 1 化合物2与ALK激酶分子对接图 |
用分子模拟方法研究了化合物2与ALK激酶的结合特征, 以揭示2在ALK活性部位的定位和取向, 以及各个基团和片段的结合模式, 指导新化合物的设计。由于当时尚未解析ALK激酶的三维结构, 故采用同源模建方法, 根据同源性强的已知InsR的三维结构, 构建了ALK三维结构。分子对接显示, 化合物2占据的位置是ATP结合腔, 结构中2-氨基嘧啶片段通过两个氢键结合固定于ALK的铰链处, 2并没有延伸到变构区的疏水腔中。皆知, Abl特异性抑制剂伊马替尼与激酶的结合位点是ATP旁边的变构区的疏水腔, 与激酶的非活性“DFG-out”构象结合, 因而呈现出比结合于ATP位点的抑制剂具有更高的选择性活性 (Schindler T, Bornmann W, Pellicena P, et al. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science, 2000, 289: 1938-1942)。然而, 化合物2结合于ATP位点却显示有高选择性抑制活性, 推测可能是由于在苯胺片段的2位存在甲氧基, 该甲氧基处于ALK铰链处的Leu258和Met259残基侧链构成的疏水性沟槽内, 而其他激酶的相应氨基酸尺寸都比较大, 位阻效应阻止了化合物2的甲氧基进入。分子模拟揭示了甲氧基的重要性 (Galkin AV, Melnick JS, Kim SJ, et al. Identification of NVP-TAE684, apotent, selective, and efficacious inhibitor of NPM-ALK. Proc Natl Acad Sci USA, 2007, 104: 270-275)。
4 先导化合物的优化 4.1 甲氧基的变换分子模拟揭示了苯胺环上的2-甲氧基对于选择性作用的重要性, 首先考察其他的烷氧基对活性的影响。在合成的化合物中, 2-异丙氧基化合物3仍保持高活性和选择性 (疏水性沟槽可容纳异丙氧基), 同时还降低了氧化代谢的程度, 表明2-异丙氧基是一个可替换甲氧基的优选基团。
4.2 消除警戒结构化合物2对体外ALK激酶、高表达ALK的细胞以及体内接种Karpas-299-或Ba/F3 NPM-ALK-细胞的小鼠都有强效抑制活性, 药代动力学性质也可以, 但却不能作为候选药物。这是因为2在体内发生氧化代谢, 产生有反应活性的代谢产物, 2与肝微粒体温孵, LC-MS证明有20%原药转化为有反应活性的亲电性物质, 例如可被谷胱甘肽捕获生成加合物 (图 2)。

|
图 2 化合物2氧化产物和与谷胱甘肽 (GSH) 的加合物 |
亲电性物质源于分子中的对苯二胺结构 (称作警戒结构, structural alert), 对苯二胺有较高的电荷密度, 是细胞色素P450的氧化位点, 生成带有正电荷的1, 4亚胺醌, 为强亲电性基团, 容易同体内亲核基团发生亲核取代反应, 具有产生特质性药物毒性 (ITD) 的风险 (Orhan H, Vermeulen NPE. Conventional and novel approaches in generating and characterization of reactive intermediates from drugs/drug candidates. Curr Drug Metab, 2011, 12: 383-394)。
4.3 变换哌嗪为哌啶环氧化代谢生成亲电性基团的原因是分子中存在的对苯二胺片段, 生成具有正电荷的亚胺醌式结构易于结合亲核基团, 从而恢复成稳定的芳香系统, 所以是强亲电性基团。氮原子含有未偶电子对, 电荷密度高于sp3杂化碳原子, 因而容易被氧化成亲电性醌式结构。为此, 将通式3的哌嗪环变换成4-哌啶结构, 消除了对苯二胺的结构因素, 设计合成了有代表性的化合物4~7。由于2和3已有高活性和选择性, 不宜对骨架结构大动, 因而固定2, 6-二苯胺基嘧啶母核不变, 将右端的哌嗪环 (可能是助溶基团) 换成4-哌啶基, 并将异丙磺酰基 (R1)、嘧啶环上的R2和苯环的R3加以变换, 有代表性的化合物3~7结构与活性列于表 1中。

|
| 表 1 化合物2~7的活性和选择性 |
表 1的数据提示, 嘧啶环上的R2为氯原子或甲基对活性和选择性有相同的影响, 苯环上R3为甲基取代可提高对人癌细胞Karpas299的活性。
4.4 哌啶环N-取代基的变换将左端的连接磺酰基的基团固定为异丙基 (变换为甲基、氨基或环丁基的化合物不如异丙基, 故被优选), 右侧苯环上固定为甲基取代, 变换嘧啶环的取代 (氯和甲基) 和哌啶N-取代基, 合成一系列化合物, 有代表性的化合物8~12的结构与活性列于表 2。
| 表 2 化合物8~12的结构、活性和选择性 |
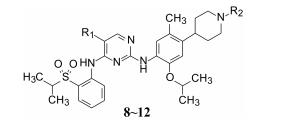
从表 2的数据可以看出, 除化合物10的活性和选择性较低外, 其余化合物都具有较高的活性和选择性。哌啶的N-取代基的极性或非极性, 基团大小的变换对活性没有显著影响, 提示氮原子和相连的基团没有与酶蛋白结合。
5 良好成药性的选择 5.1 评价化合物产生亲电性基团的警戒结构安全性是成药性的前提, 应摒弃有潜在毒性的化合物。为此, 评价了上述高活性和选择性化合物发生代谢氧化而产生亲电性基团的化合物, 方法是将受试化合物与肝微粒体温孵, 加入NADPH、谷胱甘肽 (GSH) 和尿苷-5'-二磷酸 (UDPGA), 反应后, 用LC-MS检测反应液中生成加合物的含量, 并换算成受试物的百分含量。表 3列出了有代表性化合物的数据, 表明含有对苯二胺结构的化合物2 (存在哌嗪片段) 有20%转化成亲电性物质, 与GSH生成加合物, 而含有吡啶、哌啶的化合物 (4、8、10和12) 没有发生代谢转化, 未见形成与GSH的加合物。
| 表 3 代表性化合物与GSH生成加合物的百分率 |
对有代表性的高活性化合物进行了药代动力学性质的测定, 包括在啮齿动物和人肝微粒体的清除率 (CL), 对重要的药物代谢酶CYP3A4的抑制作用 (以咪达唑仑为底物)、化合物的溶解度以及对hERG的抑制作用 (采用多非利特竞争性结合试验)。结果表明, 这些化合物对肝微粒体都有较好的稳定性; 对CYP3A4的抑制作用尚可; 有不同的水溶解度, 发现与碱性氮原子的pKa相关; 对心肌钾通道hERG的作用变化也较大, 这也与化合物的pKa相关。对化合物4和12进一步用膜片钳方法测定对hERG抑制作用的IC50值, 分别为46和20 μmol·L-1, 预示4和12对心脏是安全的。表 4列出了这些化合物的药代数据。
| 表 4 代表性化合物的药代动力学性质。*括弧内数据是用膜片钳方法测定的IC50值 |
综合体外对酶和细胞的活性和选择性、药代、安全性和物化性质, 确定了化合物4为候选药物, 进一步测定4对其他30多种激酶的的抑制活性, 结果显示, IC50低于100 nmol·L-1的激酶只有3个: IGF-IR、Ins-R和STK22D, IC50分别为8、7和23 nmol·L-1, 由于对ALK激酶的IC50为2 nmol·L-1, 选择性范围为70~230倍。对18种激酶高表达的Ba/F3细胞增殖试验表明化合物4的IC50都高于400 nmol·L-1的浓度, 而对作用靶标EML4-ALK和Tel-ALK高表达的Ba/F3细胞的IC50值分别为2.2和40.7 nmol·L-1, 显示了对细胞的高选择性。化合物4对小鼠、大鼠、犬和猴的药代动力学试验表明, 口服生物利用度F (%) 分别为55、60、100、56;静脉注射的清除率CL (mL·min-1·mg-1) 分别为27、37、9、13;半衰期t1/2 (h) 为6、9、21和26, 提示在不同动物种属之间也有良好的药代参数。进而对裸鼠移植性间变性大细胞淋巴瘤 (ALCL) 和非小细胞肺癌 (NSCLC) 作治疗性实验, 证明在可耐受剂量下有显著抑制生长作用。遂进入临床研究, 命名化合物4为色瑞替尼 (ceritinib), 经临床研究后, FDA认为对于间变性淋巴瘤激酶 (ALK) 阳性转移、经克唑替尼治疗无效的NSCLC肺癌患者, 作为突破性治疗药物, 于2014年批准上市 (Marsilje TH, Pei W, Chen B, et al. Synthesis, struc ture-activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl) phenyl)-N4 (2-(isopropylsulfonyl) phenyl) pyrimidine-2, 4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J Med Chem, 2013, 56: 5675-5690)。
诺华研制的色瑞替尼是继辉瑞于2011年上市的克唑替尼的“第二代”产品, 但它不是模仿跟进性药物。克唑替尼是个性化治疗罕见病药物的重大突破, 而色瑞替尼则是针对发生耐药和不能耐受的患者, 所以具有鲜明的创新性。
7 色瑞替尼与ALK激酶的分子对接化合物2与ALK激酶复合物的单晶结构于2010年解析 (Bossi RT, Saccardo MB, Ardini E, et al. Crystal structures of anaplastic lymphoma kinase in complex with ATP competitive inhibitors. Biochemistry, 2010, 49: 6813-6825), 图 3是将色瑞替尼分子对接到ALK晶体结构中的示意图, 嘧啶的氮原子和氨基与Met1199的氮和氧原子分别形成氢键 (图中未显示出氨基与氧形成的氢键, 虽然距离很近, 但未在同一平面之故, 若在溶液中会因柔性构象能够结合); 嘧啶环夹在Ala1148和Leu1256中间, 氯原子在疏水腔后面, 与门户氨基酸Leu1196发生疏水相互作用; 与磺酰基相连的异丙基向下弯入到由Arg1253、Asn1254、Cys1255、Leu1256、Gly1260和Asp1270组成的疏水腔内; 异丙氧基则进入由Arg1120和Glu1132的侧链以及铰链的氨基酸残基Leu1198-Ala1200-Gly1201-Gly1202组成的疏水腔。

|
图 3 色瑞替尼与ALK激酶的分子对接图 |