 2017, Vol. 52
2017, Vol. 52

编者按:
新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
利奈唑胺如同磺胺和喹诺酮类抗菌药物, 是源于有机合成的化合物。从20世纪70年代发现苗头化合物, 到2000年上市历时20余年, 杜邦和Upjohn (后为辉瑞) 公司在苗头演化成先导物和先导物的优化中, 进行了高密度的结构变换, 充分展现了药物化学、有机合成和构效关系研究的潜力, 在“众里寻他千百度”的多维探索中, 折射出首创药物的艰巨性。
1 引言抗感染药物一向是新药研发的重要领域。20世纪30年代Domagk发现百浪多息(prontosil) 开创了现代意义的化学治疗领域。20世纪40年代Fleming发现了青霉素, 形成了抗生素的治疗和半合成抗生素类药物。20世纪80年代喹诺酮类药物的诞生, 对不断出现的耐药菌的感染增添了新的治疗手段。但自此以后的20年人们期待着新型抗菌药物的出现, 所以, 当2000年以利奈唑胺为代表的噁唑烷酮药物的诞生, 作为新的作用靶标和药物类型, 增添了新的武器。
美国杜邦公司研发农作物病害药物, 报道了一组5-卤甲基-3-芳基-2-噁唑烷酮化合物具有抗作物感染的活性, 通过离体随机筛选发现化合物1 (S1623) 具有抑菌活性, 对金葡菌的最低抑菌浓度MIC=22 μg·mL-1, 对肺炎球菌的MIC=7.7 μg·mL-1 (Daly JS, Eliopoulos GM, Willey S, et al. Mechanism of action and in vitro and in vivo activities of S-6123, a new oxazolidinone compound. Antimicrob Agents Chemother, 1988, 32: 1341-1346)。虽然只有弱抑菌作用, 但因其新颖的结构类型, 公司决定进一步研究。遂以通式2为骨架, 对苯环上取代基A和甲基取代的基团B作系统变换, 进行结构优化。
2 初步优化提高了活性 2.1 噁唑烷酮环上取代基B的变换首先将A固定为乙酰基, 变换基团B, 用金葡菌(SFCO-1a) 和粪肠球菌(STCO-19) 评价化合物的最低抑菌浓度(MIC), 发现化合物3 (S构型) 活性最强, 对两种球菌的MIC分别为0.5和1.0 μg·mL-1, 与万古霉素活性相当(MIC分别为0.5和2.0 μg·mL-1), 而其他取代的化合物活性下降1~2个数量级。

|
药物分子结构中某一位置的基团对活性的贡献, 往往会因另一位置的基团变化而发生改变, 基团的活性贡献也不是一成不变的, 因而对通式2的A和B作同时的变换, 考察极性、亲脂性的不同配置对抑菌活性的影响。表 1列出了A和B不同取代化合物的对SFCO-1a和STCO-19的MIC值。
| 表 1 变换A和B对金葡菌(SFCO-1a) 和粪肠球菌(STCO-19) 的抑制作用(MIC/μg·mL-1)。*未合成; 表中数据前为SFCO-1a;后为STCO-19 |

表 1提示, 无论A为亲脂性或是亲水性基团, B只要不是乙酰氨基, 其他原子或基团取代活性都较弱或很弱, 而当B为乙酰氨基时, 除A为氨磺酰基活性较弱外, 其余的化合物都有活性。A=H2NSO2、B=NHCOCH3化合物活性低的原因可能是过强的亲水性(H2NSO2的疏水常数π=-1.82, NHCOCH3的π=-1.94)。同样A和B均为亲脂性也呈现低活性。
2.3 酰胺片段的优化化合物3显示活性最高, 但并未考察B的其他酰基对活性的影响。为此合成了有代表性的化合物4~12, 对两种菌株的活性列于表 2。
| 表 2 化合物3~12的抗菌活性 |
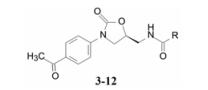
表 2的构效关系表明, 化合物3活性仍为最高, 体积小的H或大于CH3的活性都低于3。这样, 通过A和B两个位置的结构变换, 确定化合物3是优选的化合物(Gregory WA, Brittelli DR, Wang CL, et al. Antibacterials. Synthesis and structure-activity studies of 3-aryl-2-oxooxazolidines. 1. The “B” group. J Med Chem, 1989, 32: 1673-1681)。
化合物3的公司代码是Dup-721, 对包括耐甲氧西林金葡菌(MRSA) 在内的革兰阳性菌、革兰阴性厌氧菌以及结核杆菌等都有强效抑制作用。经临床前研究后, 进入临床试验。但后来发现3在小鼠的长期安全性实验中显示肝脏毒性, 因而中止了试验。
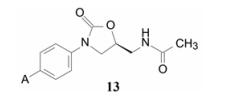
2.4 苯环上A片段的优化虽然在优化B片段时对苯环上的A片段进行过乙酰基变换, 但没有作系统的优化研究, 为此, 对通式13的A做进一步变换。
|
考察A=H、C1~C4烷基、乙烯基和乙炔基对活性的影响, 表 3显示无取代的化合物14没有活性, 碳原子数增加, 亲脂性提高, 活性提高, 正丙基(17) 和叔丁基(20) 活性最强。
| 表 3 A为不同烷基的化合物结构与活性 |
变换A为不同酰基的化合物23~26, 与3的抗菌活性比较结果列于表 4, 23是苯甲醛类化合物, 显然化学活性过强, 成药性较低。分析构效关系, 提示随着亲脂性增加, 活性提高。
| 表 4 A为不同酰基的化合物结构与活性 |
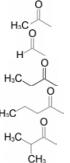
将上述的酰基还原成羟烷基, 抗菌活性显著低于相应的酰基化合物(数据从略), 推论A片段与苯环呈共轭的羰基对活性是有利的。
2.4.4 A为其他原子或基团的活性A变换为卤素、硝基、氨基、烷胺基、烷氧基、苯氧基、氰基等化合物的活性变化规律不明显, 亲脂性较强的基团活性稍高, 但总体活性偏低。
2.4.5 A为含硫的片段合成了硫醚、亚砜和砜类化合物, 甲硫醚、甲基亚砜和甲基砜有中等活性, 增大烷基活性降低。甲基砜的立体结构与叔丁基有相似性, 活性也相近。甲基亚砜为手性基团, R构型强于S构型(Gregory WA, Brittelli DR, Wang CL, et al. Antibacterials. Synthesis and structure-activity studies of 3-aryl-2-oxooxazolidinones. 2. The " A" Group. J Med Chem, 1990, 33: 2569-2578)。
2.4.6 苯环双取代对活性的影响前述的A基团变换只限于单取代基。在环上的不同位置作双取代, 构效关系表明3, 4-双取代的活性比其他位置的组合高活性的概率较高, 因此集中于这两个位置的基团变换。由于4-乙酰基已证明是优化的片段, 因而考察3-取代-4-乙酰基化合物的构效关系。4-乙酰基对活性有重要贡献(没有4-乙酰基、只有3位取代的化合物活性很弱), 而同时在3位有较小取代基的活性与无3-取代基化合物3相近, 在电性方面, 无论是推或拉电子基团都可呈现高活性。基团的尺寸对活性影响显著, 超过氯原子的临界尺寸(Es=-0.97, Es绝对值越大, 体积越大) 活性降低。所以, 3位为乙基或大于乙基的基团, 化合物都会失去活性(Park CH, Brittelli DR, Wang CLJ, et al. Antibacterials. Synthesis and structure-activity studies of 3-aryl-toxooxazolidines. 4. Multiply-substituted aryl derivatives. J Med Chem, 1992, 35: 1156-1165)。
2.5 苯环连接䓬酮的变换具有吡啶联苯结构的化合物27也显示抗菌活性, 将化合物3的乙酰基与吡啶融合, 形成既包含了乙酰基, 也有含氮芳香体系的䓬酮结构。由于䓬酮结构的不对称性, 苯环与其不同的位置连接成区域异构物28a、28b和28c。表 5列出了化合物28~33的结构与活性。
| 表 5 化合物28~33的结构与活性 |
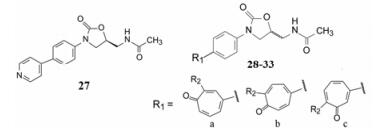
在䓬酮环上的甲氧基取代, 显示出局域异构的活性差异, 28a对两种革兰阳性菌的活性低于28b和28c大约16~32倍, 28b和28c的活性相近。同样烯丙胺29b和29c的活性也相同。化合物31c和33b对金葡菌感染小鼠的ED50与万可霉素剂量相近, S构型的化合物因是优映体, 剂量会更低(Barbachyn MR, Toops DS, Ulanowicz DA, et al. Synthesis and antibac terial activity of new tropolonesubstituted phenyloxa zolidinone antibacterial agens, 1. Identification of leads and importance of the tropone substitution pattern. Bioorg Med Chem Lett, 1996, 6: 1003-1008)。
2.6 苯环上氟原子的重要性在䓬酮系列的苯环上引入吸电子取代基三氟甲基或氯原子导致活性降低, 而且体积大的三氟甲基活性很低。而连接1或2个氟原子, 体外对耐药菌的抑制活性与无取代的相同, 但体内活性显著提高, 两个氟取代的体内活性强于单氟取代, 接近于万古霉素的体内抑菌活性。
进而考察䓬酮环的不同取代和苯环的氟代(以优映体为载体), 评价体内外抗菌活性, 䓬酮环上甲氧基取代, 苯环是一氟或二氟取代的S-异构体的活性是消旋体的2倍, 提示S构型为优映体, R构型无活性(Barbachyn MR, Toops DS, GregKC, et al. Synthesis and antibacterial activity of phenyloxazolidinone anti bacterial agens. 2. Modification of the phenyl ring -the potentianting effect of fluorine substitution on in vivo activity. Bioorg Med Chem Lett, 1996, 6: 1009-1014)。
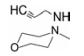
3 苯环连接含氮杂环 3.1 哌嗪和吗啉环在探索不同片段连接苯环中, 杜邦公司发现了两个芳杂环化合物, 一是前已提及的吡啶化合物27, 另一是二氢吲哚化合物34, 由于它们具有较强的抗菌活性, 而且氮原子所处的位置不同, 研究者推想哌嗪环连接在苯环上可能因模拟了吡啶和吲哚环氮原子位置而活性较强(这种推论在药物设计中比较少见, 比较勉强), 因而设计合成了化合物35和36。
化合物35特别是36 (U-100592) 综合了以前结构优化的诸多因素, 包括连接在噁唑烷酮的5位(S)-乙酰胺甲基侧链、苯环上3位氟取代、4位环取代以及N-羟乙酰化侧链等。进一步将哌嗪环变换成吗啉环, 合成了化合物37 (U-100766), 是两个高活性的里程碑式的化合物, 体外活性(MIC) 与万古霉素相当, 而且对结核杆菌有强效抑制作用, 高于异烟肼。然而36和37对大肠杆菌和肺炎克雷白杆菌等革兰阴性菌的活性很低, MIC > 64 μg·mL-1。
|
用金葡菌或肠粪球菌致死性感染小鼠, 灌胃或皮下注射化合物36和37, 其抗菌活性与万古霉素相当, 而且对万古霉素耐药菌株也有治疗效果。36和37分别命名为艾培唑胺(eperezolid) 和利奈唑胺(linezolid) 进入临床前或临床研究(Brickner SJ, Hutchinson DK, Barbachyn MR, et al. Synthesis and antibacterial activity of U-100592 and U-100766, two oxazolidinone antibacterial agents for the potential treatment of multidrug-resistant Gram-positive bacterial infections. J Med Chem, 1996, 39: 673-679)。
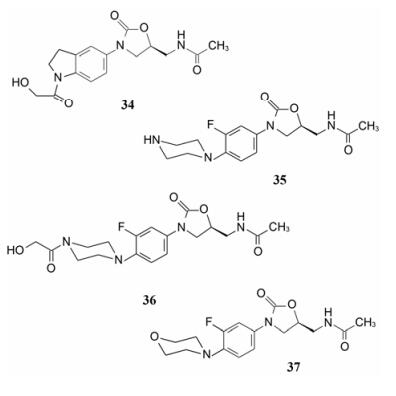
3.2 连接有吡啶、二嗪和三嗪的哌嗪噁唑烷酮化合物既然36和37的体内外抗菌活性与万古霉素相当, 促使进一步探索哌嗪环连接吡啶、吡嗪、嘧啶和三嗪等芳杂环对活性的影响, 报道了30余个有代表性的化合物, 其中化合物38~41的体内外抗菌活性与36和37相近(Tucker JA, Allwine DA, Grega KC, et al. Piperazinyl oxazolidinone antibacterial agents containing a pyridine, diazene, or triazene heteroaro matic ring. J Med Chem, 1998, 41: 3727-3735)。
|
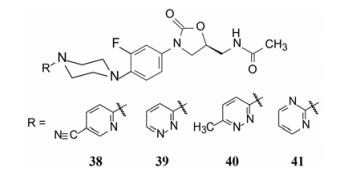
含有吗啉环的化合物37具有高抗菌活性, 自然想到合成其电子等排体硫代吗啉(42) 以及氧化物亚砜(43) 和砜(44)。有意义的是化合物42、43和44对结核杆菌H37Rv显示有高活性, MIC值都≤0.125 μg·mL-1 (异烟肼的MIC=0.2 μg·mL-1)。在动物体内化合物42被代谢优化成亚砜和砜, 而且砜化合物44与42之间没有交叉耐药现象(Michael R, Barbachyn MR, Hutchinson DK, et al. Identification of a novel oxazolidinone (U-100480) with potent antimycobacte rial ctivity. J Med Chem, 1996, 39: 680-685)。
|
化合物3、36和37都显示体内外高抗菌活性, 结构中苯环与噁唑烷酮环由单键相连, 两个环的扭角决定了分子的构象, 影响活性。为了考察分子的活性构象, 对分子骨架作限制性连接, 即将噁唑烷酮环的4'位与苯环的2位经亚甲基或二亚甲基连接, 成为通式45的化合物, 式中n=1为三环[5,6,5]并合, n=2为[6,6,5]并合; R为乙酰基、吗啉基、砜基、氨磺酰基、取代的苯基、吡啶基、嘧啶基等。由于环的并合产生了另一个手性中心, 因而两个手性碳可有4个立体异构体。报道的20余个有代表性的化合物是(±)-顺式或(±)-反式的消旋物。
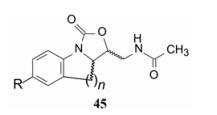
|
抗菌活性与化学结构的关系表明, 所有的(±)-顺式化合物都无活性, (±)-反式体中, [5,6,5]的活性比相应取代的[6,6,5]结构的活性高, 有些化合物可达到未并合化合物的活性。为了证明顺反异构的活性差异, 用分子模拟方法进行了研究。
分子力学(MM2) 计算了化合物3的最低能量构象群的苯环与噁唑烷酮环的扭角平均为-29°, 有活性的反式化合物(R=CH3CO, n=1) 的扭角为-24°, 而无活性的顺式体(R=CH3CO, n=1) 扭角为+21°, 说明未并合为三环的化合物(如化合物3、36和37) 与细菌靶标是以类似于反式构象的负性扭角相结合的(Gleave DM, Brickner SJ, Mannine PR, et al. Synthesis and antibacterial activity of [5,6,5] and [6,6,5] tricyclic oxazlidinones. Bioorg Med Chem Lett, 1998, 8: 1231-1236)。
5 候选药物的选定和利奈唑胺上市上述研发噁唑烷酮类新型抗菌药物的过程, 呈现出数个体内外抗菌活性高、抗菌谱广的里程碑式化合物, 例如化合物3 (代号DuP-721)、36 (艾培唑胺)、37 (利奈唑胺) 和42 (代号U-100480) 等, 经动物的安全性评价和药代动力学性质比较, 利奈唑胺优胜于其他候选物, 37的口服生物利用度F=100%, 血药浓度几乎与静脉注射相同, 口服1~2 h后血浆浓度达峰, 与血浆蛋白结合率约为30%, 血浆半衰期t1/2=5 h, 遂进入了临床研究, 经三期临床试验证明对革兰阳性菌感染的患者有显著治疗作用, 于2000年经FDA批准由辉瑞公司上市。
6 利奈唑胺的作用机制和结合特征利奈唑胺是细菌蛋白合成的抑制剂, 作用环节是阻断核糖体上mRNA翻译成蛋白的过程。虽然该过程还不完全清楚, 但已证明利奈唑胺作用于蛋白合成的起始阶段, 而不是像大多数蛋白抑制剂作用于链延长阶段。利奈唑胺的结合位点是核糖体50S亚基的A位点, 即肽转移酶中心(PTC), PTC是肽键形成的位置(图 1)。通过解析利奈唑胺与嗜盐古生菌的S50复合物晶体结构, 表明当利奈唑胺结合于细菌S50的A位点之后, 进入到由RNA构成的PTC的腔, 改变了一些重要核苷酸的构象, 例如U2539的位置改变, 使尿嘧啶环与噁唑烷酮环发生范德华作用; C2487移动, 胞嘧啶环与苯环发生π-π堆积作用; 酰胺侧链的氨基与G2540的磷酸形成氢键等, 由于利奈唑胺的结合, 阻止了氨基酸酰化的tRNA进入A位点, 并最终使tRNA离开核糖体。图 2是利奈唑胺与S50的PTC复合物晶体结构图(Ippolito JA, Kanyo ZF, Wang DP, et al. Crystal structure of the oxazolidinone antibiotic linezolid bound to the 50S ribosomal subunit.J Med Chem, 2008, 51: 3353-3356)。

|
图 1 细菌核糖体合成蛋白的示意图 |

|
图 2 利奈唑胺与细菌核糖体S50的晶体结构 |
首创药物利奈唑胺于2000年上市, 第二个被FDA批准的特地唑胺(46, tedizolid) 是2014年由Cubist上市的同类药物, 间隔了14年之空白, 这与1986年首创的诺氟沙星之后, 迅速有多种喹诺酮类药物的跟随上市, 形成鲜明差异。这可能与首创者对噁唑烷酮的结构优化比较充分、难以突破知识产权有关, 可从本文叙述的内容反映出来。特地唑胺是以磷酸酯为前药, 对耐受甲氧西林的革兰阳性菌的抑制作用强于利奈唑胺4~16倍。可口服或注射用药, 日用一次(Schaadt R, Sweeney D, Shinabarger D, et al.In vitro activity of TR-700, the active ingredient of the antibac terial prodrug TR-701, a novel oxazolidinone. Antim icrob Agents Chemother, 2009, 53, 8: 3236-3239)。