 2016, Vol. 51
2016, Vol. 51

1998年美国霍华德休斯医学研究中心(HHMI)发现了一种肽类神经递质, 命名为下视丘泌素(hypocretin) (De Lecea L, Kilduff TS, Peyron C, et al. Proc Natl Acad Sci U S A, 1998, 95: 322-327), 是源于在下丘脑(hypothalamus)发现了特异性的mRNA。同年Scrips研究所也独立发现了一种神经递质, 因为可激活食欲, 称之为食欲素(orexin) (Sakurai T, Amemiya A, Ishii M, et al. Cell, 1998, 92: 573-585)。其实, 他们发现的是同一种内源性的肽类物质。食欲素的主要功能是调节食物摄取, 高表达食欲素的小鼠可增加更多的体重。因而引起制药界的注意, 希望以食欲素为切入点研究减肥药。
然而Lin等于1999年发现orexin与睡眠有关(Lin L, Faraco J, Li R, et al. Cell, 1999, 98: 365-376)。通过遗传学和药理学实验证明orexin与已经知晓的G蛋白偶联受体结合, 即orexin与Ox1R和Ox2R结合后, 引发的信号通路与周期性的睡眠和苏醒有密切关联。这样, 一度曾以orexin及其受体作靶标、研究减肥药为目标的公司纷纷转向催眠药的研究。
2 靶标和活性评价Orexin为两种神经肽orexin A和orexin B的总称, 广泛分布于大脑中, 调控睡眠与觉醒过程。Orexin的受体有两种亚型: Ox1R和Ox2R。Ox1R与orexin A的亲和力强于orexin B, 而Ox2R对orexin A和B的结合力是相同的。Orexin与受体结合的生理功能在调节睡眠-苏醒循环过程起主导作用。敲除orexin基因的小鼠的生理特征与人的发作性睡病相同, 白昼长时嗜睡, 发生快速眼动睡眠(REM sleep)。因此, Orexin受体作为药物靶标, 它的抑制剂可成为失眠的治疗药; 反之, 如果能够激活orexin受体, 又可治疗睡眠病。Orexin及其受体作为新的作用环节, 意味着可发现新型药物。
为了进行体外通量筛选, 默克公司构建了两种评价化合物活性的模型:一是用放射性同位素标记的底物orexin与人的Ox1R和Ox2R受体结合, 用置换实验测定化合物对受体的亲和力, 本文中用Ki表示; 另一是用荧光成像涉猎仪(fluorometric imaging plate reader system, FLIPR)测定对细胞的作用。用CHO细胞表达的人Ox2R (hOx2R)和发生Ile408Val变异的人Ox1R (hOx1R)两株细胞, 分别测定化合物抑制hOx2R和hOx1R的功能, 用IC50表示。研发的化合物目标是对这两种受体亚型和两株细胞分别有高亲和力和抑制活性。
3 苗头化合物及其向先导物的过渡(随机筛选获得苗头化合物)默克用公司的化合物库(300万个化合物)对hOx2R和hOx1R的活性进行了随机筛选, 发现化合物1有良好的活性, 对hOx1R的活性Ki=150 nmol·L-1、IC50=630 nmol·L-1, 对hOx2R的活性Ki=5 nmol·L-1、IC50=98 nmol·L-1。1对hOx1R的活性明显较弱, 优化结构使对二者的活性相近。
由苗头过渡到先导物的结构变换, 宗旨是保持1, 4-二氮䓬母核不变, 变换的是母核左侧的杂环和右侧的苯甲酰片段。
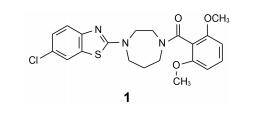 |
去除化合物1中的氯原子, 2的活性有所下降, 同样没有取代的苯并噁唑化合物3的活性与2相近。苯并噻唑简化为噻唑环成化合物4, 活性显著降低。将苯并噻唑开环成5, 则失去活性。说明体积较大的环系有利于活性。这些SAR在后文叙述活性化合物的构象特征得到了解释。若苯并噻唑环变为喹啉(6)、喹喔啉(7)和喹唑啉(8)等含氮[6, 6]芳杂环, 活性与1相近, 但抑制细胞的活性提高, IC50值下降。这种受体结合实验的变化不大, 而对细胞的活性变化大, 符合于一般性规律:用靶标作分子水平的测定, 亲和力(活性)对结构变化比较敏感, 往往揭示构效关系; 而用细胞评价活性(抑制或激动功能)则更取决于整个分子的物化性质, 往往对结构的细微变化不敏感, 反映出穿越细胞膜和在细胞内扩散起重要影响。表 1列出了不同杂环的化合物结构及其活性。
| 表 1 变换杂环的化合物(2~8)结构及其活性 |

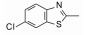

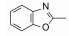
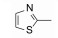
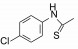
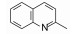
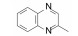
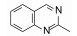
表 2列出了变换芳酰基合成的化合物结构及其活性。将1的酰基还原成CH2, 即为叔胺化合物9, 活性丧失殆尽。1的苯环上去除1个甲氧基, 化合物10活性降低。11是1的同系物苯乙酰化合物活性更为减弱。取代的苯基被甲基呋喃置换(12)也失去活性。用联苯甲酰置换的化合物13与受体的亲和力比1提高5倍, 但对细胞的抑制活性有所降低, 分析原因可能是13的亲脂性过强(clog P=6.1), 而1的clog P=4.5, 亲脂性过强不利于溶解与扩散。为了调整亲脂/亲水性, 用有极性的基团连接苯环, 其中发现三唑化合物14 (clog P=4.1)的受体结合实验维持高活性, 并且对细胞的抑制活性也显著提高, 两株细胞的IC50分别为36 nmol·L-1和39 nmol·L-1。苯环与三唑连接的位置对活性影响较大, 例如1-三唑化合物对Ox1R和Ox2R的Ki值分别为1 050和75 nmol·L-1。在2-三唑取代的基础上连接甲基, 化合物15的活性更高。
| 表 2 变换芳酰基的化合物(9~15)结构及其活性 |
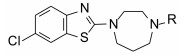
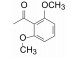
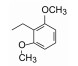
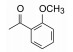
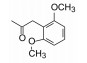
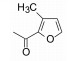
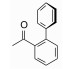
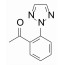
综合上述以不变的1, 4-二氮䓬母核为中心的结构优化, 将对活性贡献最大的片段组合在一起, 设计合成了对Ox1R和Ox2R受体有高活性的双重拮抗剂16。由于Ox1R和Ox2R是研发睡眠药的新靶标, 化合物16应显示体内效果, 即对实验动物应有催眠作用。通过受体水平、细胞水平和动物实验, 对靶标和化合物双方的成药性进行概念验证。
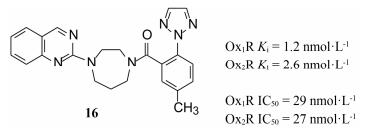 |
为证明16能够阻断脑中orexin-受体的信号通路, 测定了化合物对大鼠脑切片的中缝背核中的神经元放电频率的调节作用, 结果表明16能够抑制orexin A引起的神经元放电频率, 并且抑制强度与给药浓度呈正相关性。化合物16灌胃大鼠的体内实验表明, 可抑制orexin信号系统, 减少了大鼠的清醒时间。并在大鼠的各种状态下, 记录皮层脑电图和心电图, 都表明该双重抑制剂促进了睡眠过程。
化合物16的clog P=3.1, 实测log P=2.9, 可形成盐酸盐以提高溶解性, 而且有良好的过膜性(Papp=38×10-6 cm·s-1)。然而, 大鼠灌胃16的生物利用度很低, F=2%, 半衰期很短, t1/2=21 min。对犬的F=16%, t1/2=1.28 h, 这些较差的药代性质未能达到成药性的要求, 需要在化合物16的基础上进一步优化(Whitman DB, Cox CD, Breslin MJ, et al. ChemMedChem, 2009, 4: 1069-1074)。
5 第二轮的优化-改善药代动力学性质(从研究代谢产物入手)化合物16生物利用度低的主要原因是有首过效应的缘故, 在肝脏中发生氧化代谢而失活。通过16与微粒体温孵, 温孵液的氧化产物(往往是醛酮类化合物)用氨基脲捕获羰基化合物, 质谱分析加成物, 得知是二氮䓬的7位被氧化成羟基, 开环成醛基而形成的缩氨脲。二氮䓬开环, 打散了16的刚性结构。苯环上的甲基也可被氧化, 以及喹唑啉的氧化等。如图 1所示的代谢位点。
 | 图 1 化合物16被氧化代谢的位点 |
阻止氧化代谢的一个方法是在代谢位点附近引入基团以增加位阻, 为此在二氮䓬的不同位置作不同的取代。
首先, 成功完成了不同取代的二氮䓬环的合成, 共制备了15个单取代和二取代的二氮䓬母核, 通过通量的合成和活性测试, 证明2、6或7位引入基团不仅不影响与受体的结合, 而且还提高了活性强度。将这些有潜在活性的母核与不同的杂环和(或)不同取代的苯甲酸缩合, 得到了大量目标化合物。生物学评价分3步走:首先测定对双靶标Ox1R/Ox2R的活性, 然后犬静脉注射测定清除率以评价代谢速率和程度, 在此基础上评价口服生物利用度。综合评价化合物的活性、药代和物化性质, 确定了单取代的7-甲基二氮䓬为母核, 做进一步优化。
5.2 喹唑啉环上的取代在合成的化合物中, 有代表性的化合物17~19对两种Ox1R/Ox2R受体的结合力与抑制活性都很高, 然而犬的药代实验表明, 化合物17和18的药代性质与16相比没有明显改善, 而6-氟代喹唑啉化合物19的生物利用度和清除率都优于16, 灌胃引起大鼠同样睡眠的剂量只是化合物16的十分之一, 药效高是因药代好之故。表 3列出了这些化合物的活性和犬的药代数据。
| 表 3 有代表性的化合物的活性和犬的药代数据 |
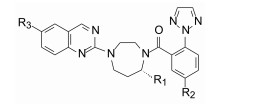
然而化合物19出现新的问题, 即在体内经氧化代谢生成亲电性基团, 有潜在的毒性。用微粒体与19温孵, 加入谷胱甘肽(GSH)以捕获亲电试剂, 仪器检测到了GSH加成产物。药物代谢产生亲电性物质, 往往引起特质性毒性反应, 因而化合物19不能作为候选化合物。因此优化的目标又加上了是否会因代谢而产生有潜在毒性的亲电性分子。研究者指出, 亲电性基团的结构片段是氟代喹唑啉环所产生, 所以变换结构集中于左侧的杂环处, 为此, 将7-甲基二氮䓬和三唑基苯甲酰片段固定不变。
5.3 杂环的优化和苏沃雷生的诞生这个阶段的优化目标, 是在保持或提升对双靶标的抑制活性、具有良好的药代性质的同时, 还要避免或降低在体内代谢而产生亲电性物质的风险, 优化的结构部位是左侧的杂环。评价目标化合物潜在的特质性毒性的方法, 是将受试物与人肝微粒体温孵, 温孵液中放有足够量的GSH (谷胱甘肽), 以捕获氧化代谢生成的亲电物质, 所有生成的GSH加成物, 用LC-MS测定并计算GSH加成物的峰面积, 与测定液中预加的内标物(IS)峰面积的比值是化合物产生亲电基团的程度, 比值小表明安全度高。
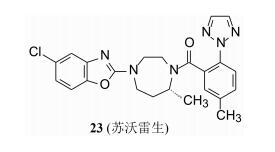
为降低喹唑啉环的被氧化趋势, 引入第2个氟原子以降低芳环的电荷密度, 二氟代物20的活性和药代与基准物19相同, 但代谢活化反而强于19, 提示喹唑啉环需要撤换(决策果断)。合成的四氢喹唑啉化合物21活性未变, 代谢活化也较低, 但犬的清除过快, 药代不适。研究者想到早期优化制备含有苯并噁唑的化合物的药代清除率较低, 因而合成了22, 其清除率果然低于19。然而它与Ox1R/Ox2R受体的结合作用减弱了数十倍, 22不值得深入研究。联想到前述化合物2因去除了化合物1中的氯原子活性丧失殆尽的事实, 说明杂环部位的亲脂性对活性的重要影响, 因而合成了5-氯苯并噁唑化合物23, 从表 4中化合物23的各种数据可以看出23是个比较理想的化合物。
| 表 4 不同杂环的化合物结构和活性 |
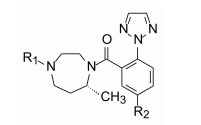





化合物23的药代动力学表明, 对犬有良好的口服生物利用度, F=56%, 有中等/较低程度的清除率, 它的代谢产物主要是苯环上甲基被氧化成羟甲基, 后经葡萄糖醛酸苷化自尿中排出。少部分的二氮䓬环上甲基氧化成羟甲基, 以及噁唑环的某处发生氧化。大鼠灌胃30 mg·kg-1化合物23, 表现出有良好的睡眠/觉醒周期。遂进入临床前和临床研究, 命名为苏沃雷生(suvorexant)。经三期临床研究表明苏沃雷生是安全有效的促睡眠药物, 于2014年经FDA批准由默克公司上市(Cox CD, Breslin MJ, Whitman DB, et al. J Med Chem, 2010, 53: 5320-5332)。
 |
Ox1R/Ox2R受体是G蛋白偶联受体, 结构尚未解析。默克公司以化合物17为模板, 用二维核磁共振、晶体X-射线衍射和分子模拟研究了17的最低能量构象。结果表明, 17和苏沃雷生的二氮䓬环上甲基为R构型, 二氮䓬的七元环呈扭船式(twist-boat)构象, 使得杂环(喹唑啉)与苯环处于顺式, 分子构象呈弯曲的U形, 苯环与喹唑啉环发生π-π叠合作用, 苯环上甲基加助了这种结合。由NMR的1H化学位移数据推断17在氘代甲醇溶液中的低能构象与分子模拟的结构相吻合, 单晶X-射线衍射图也与之相似。图 2a是用分子模拟化合物17最低能量的分子模拟图, 图 2b是单晶衍射图。该最低能量的构象应与活性的药效构象相近。
 | 图 2 化合物17的最低能量的分子模拟图(a)和单晶衍射图(b) |
虽然未曾报道苏沃雷生的最低能量构象, 但具有亲脂性的氯代苯并噁唑也应与甲基苯环形成π-π叠合, 因而与化合物17有相似的药效构象(Cox CD, McGaughey GB, Bogusky MJ, et al. Bioorg Med Chem Lett, 2009, 19: 2997-3001)。此外, 用合成的构象限制的大环化合物的构象和活性进一步证明了上述的U形构象(Coleman PJ, Schreier JD, McGaughey GB, et al. Bioorg Med Chem Lett, 2010, 20: 2311-2315)。