 2016, Vol. 51
2016, Vol. 51



2. 中国医学科学院、北京协和医学院药物研究所, 北京 100050
2. Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100050, China
JAK激酶(Janus kinase)和其下游的效应器、信号转导及转录激活蛋白(signal transducers and activators of transcription proteins, STATs)形成了重要的细胞因子信号传导途径— JAK-STAT通路[1]。该通路的发现, 大大提高了研究者对基因调控的认识[2], 研究发现JAK-STAT通路可由多种细胞因子、生长因子以及受体激活, 参与细胞增殖、分化、凋亡、血管生成以及免疫调节等过程[3]。JAK激酶是JAK-STAT信号通路中的关键激酶, 从20世纪90年代初, 首个JAK激酶被发现以来, 直到2012年, 第一种JAK激酶抑制剂(tofacitinib)才被批准用于类风湿性关节炎(rheumatoid arthritis, RA)的治疗[4]。JAK激酶在包括造血细胞、淋巴细胞在内的多种类型细胞的分化、增殖和存活中起着重要作用。JAK-3是JAK激酶家族成员之一, 相较于其他几种JAK激酶, 其主要分布于骨髓和淋巴系统中, 是免疫反应细胞信号传导的一个关键分子[5], 与免疫系统的调控密切相关, 所以针对JAK-3为靶点设计、合成小分子抑制剂, 作为治疗自身免疫疾病的药物受到广泛关注, 已有多个JAK-3激酶抑制剂进入临床研究, 表现出较好的开发前景。本文对JAK-3激酶的结构功能以及JAK-3激酶抑制剂研究状况进行综述。
1 JAK激酶的生物学 1.1 JAK激酶家族在哺乳动物体内, JAK激酶家族有4个成员: JAK-1、JAK-2、JAK-3和TYK-2, 由超过1 100个氨基酸组成, 相对分子质量可达120 000~140 000, 同源性达40%~70%, 这些JAK激酶家族成员从C端到N端依次可分为7个同源结构域(JAK homology domain, JH) (图 1): JH1为激酶区, 在JAK家族中高度保守, 用以编码激酶蛋白, 激酶结构域中活化环(activation loop)内酪氨酸的磷酸化状态决定了JAK蛋白的激酶活性[6]; JH2为激酶样区或“假”激酶区, 该假激酶结构域是JAK蛋白区别于其他酪氨酸蛋白的独特属性, 该激酶区虽不具有催化活性, 但对JH1的活性起调节作用, 该结构域内的突变常可导致JAK激酶活性的增强或者减弱, 并进而导致某些疾病的发生[7, 8]; JH3~JH4为SH2结构域(Src homology 2 domain), 该结构域含有约100个氨基酸残基, 其可以特异性地识别和结合配基上磷酸化的酪氨酸残基[6, 9]。JH5~JH7为FERM结构域, 该结构域保守, 主要调节JAK与受体的结合[10, 11]。JAK-3作为JAK激酶家族成员之一, 在结构上, 同样含有上述的激酶区, 其不同结构域内特定氨基酸的突变也会造成其激酶活性的改变[12, 13]。

|
Figure 1 Domain structure of Janus kinases (JAKs) |
JAK-STAT信号通路是多种细胞生长、活化、分化、凋亡及其功能发挥过程中重要的一条细胞内信号转导途径。STAT是一类能与靶基因调控区DNA结合的胞质蛋白, 是JAK的下游底物。STAT家族中包括STAT1、STAT2、STAT3、STAT4、STAT5A、STAT5B及STAT6等7个成员。JAKs与STATs之间的相互作用在细胞因子受体信号通路中起着重要作用[14, 15]。当细胞表面的细胞因子受体与其各自的细胞因子配体结合后引起受体分子的二聚化, 这使得与受体偶联的JAK激酶相互靠近并通过交互的酪氨酸磷酸化作用而活化。JAK激活后催化受体上的酪氨酸残基发生磷酸化修饰, 继而这些磷酸化的酪氨酸位点与周围的氨基酸序列形成“停泊位点” (docking site), 同时含有SH2结构域的STAT蛋白被招募到这个“停泊位点”。最后, 激酶JAK催化结合在受体上的STAT蛋白发生磷酸化修饰, 被激活的STAT蛋白离开受体并组成二聚体后, 再转移到细胞核内对特定的基因进行转录调节[16], 如图 2所示。

|
Figure 2 Cytokine activation of the JAK-STAT pathway[16] |
JAK-STAT信号通路是一条由多种细胞因子受体刺激的信号转导通路, JAK激酶介导细胞内大多数细胞因子的信号传导[17], 如白介素(IL)类、干扰素(IFN)类、促红细胞生成素(EPO)、粒细胞和巨噬细胞集落刺激因子(GMCSF)、促生长素(GH)、催乳素(PRL)、促血小板生成素(TPO)、血小板衍生因子(PDGF)以及表皮细胞生长因子(EGF)等, 而且不同受体可激活不同亚型的JAK激酶, 从而表现差异化的生物学功能[18-20], 如表 1所示。其中, JAK-3通过与IL-2、IL-4、IL-7、IL-9、IL-15和IL-21等I型细胞因子受体复合物中的γ链(γc)相结合, 调节细胞信号传导。当JAK-3缺陷或γc突变时, 可导致重症联合免疫缺陷(severe combined immunodeficiency, SCID)[21], 表现为T细胞和自然杀伤细胞(natural killer cell, NK)减少、B细胞功能丧失等免疫限制的症状[22]。
| Table 1 The four JAKS are used by many cytokine-receptor complexes[17]. CNTF: Ciliary neurotrophic factor; EPO: Erythropoietin; GH: Growth hormone; GM-CSF: Granulocyte macrophage-colony stimulating factor; IBD: Inflammatory bowel disease; LIF: Leukaemia inhibitory factor; LP: Leptin; OSM: Oncostatin M; PRL: Prolactin; RA: Rheumatoid arthritis; SLE: Systemic lupus erythematosus; TPO: Thrombopoietin; TSLP: Tymic stromal lymphopoietin |
JAK-1、JAK-2和TYK-2在人体各组织细胞中均有表达, JAK-3主要表达于各造血组织细胞中, 主要存在于骨髓细胞、胸腺细胞、NK细胞及活化的B淋巴细胞、T淋巴细胞中[23]。JAK-1和JAK-2等缺失会造成人体致死性损伤, JAK-3缺失则可避免损伤其他组织细胞的毒性不良反应[24, 25]。小鼠基因敲除实验研究发现, 若敲除小鼠的JAK-1和JAK-3基因都将表现出淋巴系统缺陷, 导致SCID的发生; 若敲除小鼠的JAK-2基因, 小鼠将无法存活; 而敲除TYK-2基因的小鼠可以存活, 但将导致抗菌免疫功能下降并增加了病毒感染、肺部感染的可能性, 同时也会诱导类风湿性关节炎的炎性反应[26]。
综上, 基于JAK激酶家族中各亚型的功能特点和特殊的组织分布, JAK-3已成为治疗自身免疫性疾病的热门靶标。目前, 越来越多临床研究也将RA的治疗聚焦于阻断JAK-3信号转导通路上。
2 JAK-STAT信号通路在RA发病中的作用RA是一种以关节病变为主的慢性全身自身免疫性疾病, 表现为免疫系统对关节等自身组织的持续损害, 由多种免疫细胞(B淋巴细胞、T淋巴细胞及巨噬细胞等)及相关细胞因子参与介导的复杂疾病, 目前关于RA的发病机制尚未完全明确, 随着多种激酶在免疫激活和炎症信号通路中的关键作用机制被深入认识, 选择性激酶抑制剂已被视为治疗RA的新策略[27]。
研究[28, 29]发现, 在RA发病过程中, IL-2、IL-6、IL-17、IL-21、IFNs及GM-CSF等细胞因子, 在RA滑膜细胞和滑膜组织中的水平明显升高, 这些因子可通过不同途径激活JAK-STAT信号通路, 例如: IL-2、IL-15、IL-21可与JAK-3结合。研究发现, 在RA发病过程中, STAT1和STAT3发挥了主要作用[30]。Wang等[31]发现RA滑液的单核细胞中STAT3具有显著的DNA结合活性, 并且RA滑液中的可溶性因子能有效激活STAT3。随后在动物模型上的研究结果表明, STAT3失调能够改变关节炎的炎症过程。Krause等[32]和de Hooge等[33]同样发现, STAT3在动物关节炎模型中处于持续激活状态, 诱导关节炎慢性炎症反应, 而抑制STAT3功能, 进而使EGF由促滑膜细胞生长存活的因子转变为致其死亡抑制的因子。由此可见, STAT3是RA发病过程中一个关键性致病因子, 其通过抑制成纤维细胞凋亡、促进T细胞存活及抗体产生而调控RA发病过程中滑膜细胞的异常生长和存活。通过免疫组织化学方法对RA患者的滑液的研究[34]发现, 炎症反应明显的RA患者体内STAT1的表达明显增加, 并且STAT2与STAT1及干扰素调节因子9 (interferon regulatory factor 9, IRF9)可以形成异二聚体转录复合物, 推测在RA发病过程中, STAT2与STAT1共同作用, 通过激活炎症相关基因的表达在促进滑膜炎症反应中发挥作用。然而, Krause等[32]和de Hooge等[33]却发现, STAT1还可通过诱导细胞因子信号抑制物1 (suppressor of cytokine signaling 1, SOCS1)的表达发挥抗炎作用。因此, STAT1在RA发病过程中, 可能具有双重作用, 在疾病发展的不同阶段, 分别表现出促进或抑制炎症作用。Finnegan等[35]的研究表明, IL-4可通过STAT6调控炎症。STAT作为JAK的下游底物, 其生理作用的实现均受到JAK的调控, 若选择性地抑制JAK-3的活性能够实现对STAT-3等的抑制, 进而对RA等起到治疗作用。因此, 鉴于JAK-STAT信号通路与RA发病机制有着重要的关系, 选择性JAK激酶抑制剂也被视为治疗RA新靶点。近年来, JAK靶点的研发已取得了一定的成果, 2012年, 选择性JAK-3抑制剂tofacitinib通过了临床试验, 被批准用于类风湿性关节炎的治疗。
3 JAK-3激酶抑制剂由于JAK-3在JAK-STAT信号通路中具有重要作用, 并且其仅在特定的组织中进行表达, 因而抑制JAK-3活性后导致免疫抑制但不会引起更多的非正常生理变化, 也就意味着在疾病的治疗中导致不良反应的几率更低, 这也使得JAK-3成为治疗自身免疫疾病的热门靶点。现有的激酶抑制剂多是ATP竞争性抑制剂。JAK-3的激酶域具有保守的双裂片结构, N端含有6个反向平行β折叠和1个α螺旋, 起催化调节作用。同时, 还有1个在ATP磷酸化过程中起调节作用的甘氨酸富集区(P-loop区)[36], C端主要由7个α螺旋和2个短β折叠组成, 起催化作用, 催化调节区和催化区通过一个“hinge”区连接。ATP结合位点就位于催化调节区和催化区中间, 与“hinge”区相邻。该活性腔主要由2个β折叠构成一个发夹样口袋, 袋口呈扁平状, 口袋内腔较大, 纵向跨度深, 活性位点的大小约为8 Å × 11 Å × 20 Å, 体积为530 Å3[37], 如图 3所示。ATP结构中的嘌呤环可结合于JAK-3激酶结构的疏水口袋, 并与连接的氨基酸残基形成氢键, JAK-3激酶抑制剂则可竞争性地结合ATP与JAK-3激酶结合位点。目前, 已经研发出多种结构类型的选择性JAK-3抑制剂, 包括吡咯并嘧啶类、吡咯并吡啶类、吡咯并吡嗪类和马来酰亚胺类等。

|
Figure 3 Three dimensional structure of JAK-3[37]. A: The model of JAK-3 showing the molecular surface of protein [blue] and catalytic [ATP-binding] site [yellow]; B: The model of unoccupied space in the catalytic [ATP binding] site of a JAK3 homology model |
Tofacitinib (CP690550, 1)是Pfizer公司研发的一种吡咯并嘧啶类选择性JAK-3激酶抑制剂, 2012年11月6日成为首个获美国FDA批准用于RA治疗的JAK激酶抑制剂。Tofacitinib对JAK-3的抑制活性(IC50=1 nmol·L-1)是JAK-2 (IC50=20 nmol·L-1)的20倍及JAK-1 (IC50=112 nmol·L-1)的100倍, 而对TYK-2几乎无活性[38]。Chrencik等[39]和Meyer等[40]报道对tofacitinib的立体化学结构进行研究, 发现其手性结构决定其能够特异性地结合到JAK-3分子上, 从而抑制JAK-3磷酸化, 进一步导致STAT磷酸化受阻, 造成下游炎性细胞因子合成受到抑制。Tofacitinib在临床研究中表现出良好的临床疗效, 在Ⅲ期联合治疗临床试验研究中, 797例对甲氨蝶呤(methotrexate, MTX)反应不敏感的RA患者在接受MTX的同时, 随机加用tofacitinib (5 mg和10 mg, bid, po)或等量安慰剂, 结果显示, 治疗6个月后, 5和10 mg tofacitinib组患者ACR20反应率分别为51.5%和61.8%, 与安慰剂组相比有显著统计学差异, 10 mg组患者关节肿胀改善更加明显; tofacitinib各剂量组受试者的身体功能得到明显改善[以健康调查表疾病指数(HAQDI)为评价指标][41]。同时, tofacitinib用于生物制剂治疗无效的RA患者, 仍然能产生满意的临床效果[42]。此外, 临床试验研究中也发现使用tofacitinib与严重感染风险增高相关, 其长期安全性仍有待进一步研究。目前, tofacitinib还应用于多个不同适应证的临床研究, 如干眼病、克罗恩病、银屑病、溃疡性结肠炎和器官移植等[43-46]。
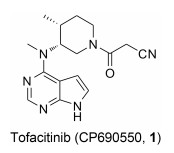
|
Peficitinib (ASPO15K, 2)是由Astellas和Johnson & Johnson公司联合开发的选择性JAK-3和JAK-1抑制剂, 其对JAK-3的IC50为0.71 nmol·L-1, 对JAK-1、JAK-2及TYK-2的IC50分别为3.9、5.0和4.8 nmol·L-1, 但是, 对IL-2介导的人T细胞的IC50为18 nmol·L-1, 在促红细胞生成素诱导白血病细胞增殖实验中, 对JAK1/JAK3的选择性约为JAK2的14倍。该药物在大鼠的AIA (adjuvant-induced arthritis)模型中表现出很好的有效性, 每日口服剂量为1~30 mg·kg-1 [47]。该药于2014年2月完成了Ⅱa期临床研究, 目前正在日本、美国和欧洲开展Ⅱb期临床研究, 其中用于日本患者的12周单药结果表明, 该药物具有很好的有效性和耐受性[48]。同时, 本品于2012年开展的一项为期6周的银屑病临床研究发现, 每天服用50 mg剂量peficitinib就可达到有效的治疗作用, 该研究证明其对中度至重度银屑病具有治疗潜力[49]。有信息表明, Astellas公司已经开始在类风湿性关节炎患者中开展peficitinib的Ⅲ期临床试验[50]。

|
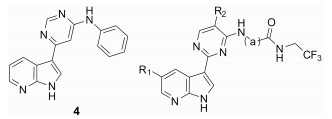
Decernotinib (VX-509, 3)是由Vertex公司开发的一种吡咯并吡啶类小分子选择性JAK-3激酶抑制剂, 该化合物于2013年完成了Ⅱb期临床研究, 临床试验发现本品兼具良好的生物活性、高选择性和耐受性[51], 与同类药物tofacitinib相比, 本品不会造成中性粒细胞减少[52]。于2013年4月, decernotinib率先在美国和爱沙尼亚进入了Ⅱ/Ⅲ期临床研究。
Decernotinib对JAK-3的Ki值为2.5 nmol·L-1, 对IL-2介导的人T细胞的IC50为0.14 μmol·L-1与tofacitinib相当(0.15 μmol·L-1), 对特异性细胞选择性在25~150倍[52], 其在体选择性优于tofacitinib。Decernotinib的开发是以化合物4为先导化合物, 在3位用一系列含氮杂环替换后, 活性显著提升(3, 5~8)。先导化合物5位用氯取代后(6~8), 化合物活性改善不大, 并且不利于成药。在大鼠药代动力学实验中, 化合物3的药物半衰期为5.1 h, 在后续一系列在体实验中, 化合物3表现出良好的成药性。Decernotinib是目前ATP竞争性JAK-3抑制剂中很有希望进入市场的药物(表 2)。
| Table 2 SAR studies for pyrrolo-pyridine analogues |



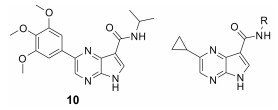
2013年, Roche公司公布了一系列3-氨基-5-环丙基吡咯并吡嗪类化合物, 其中化合物9抑制JAK-3活性为0.26 nmol·L-1, 与tofacitinib相比, 其体外激酶实验的活性要优于tofacitinib, 而对JAK-3的选择性却低于tofacitinib, 对JAK-1和JAK-2的IC50分别为3.2和0.8 nmol·L-1, 但是在细胞实验中, 化合物9对JAK-3的选择性要明显优于tofacitinib。
Roche公司通过化合物筛选发现了对JAK激酶具有较好抑制活性的化合物10, 但该化合物对JAK激酶的选择性低, 因而以化合物10作为先导化合物, 对其关键位置5位进行了多种取代基的探索, 当5位为环丙基时(11), 化合物的JAK激酶选择性提高。在此基础上, 进一步对3位进行改造, 分析化合物11与JAK-3复合物(图 4), 发现JAK-3活性腔中存在Leu956和Ala966两个氨基酸残基, 因而在化合物3位引入异丙基酰胺结构(12~14)以希望能与Leu956和Ala966形成疏水作用, 提高化合物的活性和选择性, 活性测试也验证了该猜想, 用仲丁基取代异丙基(12), 化合物的活性和选择性得到了显著提升, 当用叔丁基衍生物取代时(14), 化合物抑制JAK-3的活性首次达到1 nmol·L-1水平。为了填充活性腔“上部”的空间, 化合物首先引入了简单的双酰胺基的结构(15~17), 然而这些化合物的活性并未表现出明显的改善, 通过引入氰基, 对该类化合物进行进一步优化(9~18), 发现氰基的引入有利于提高化合物的活性。综合化合物的构效关系, 得到了最终高活性产物化合物9[53] (表 3)。

|
Figure 4 Binding mode of compound 11 within the JAK-3 binding site[53] |
| Table 3 SAR studies for 3-amido-5-cyclopropyl pyrrolopyrazine analogues |
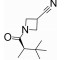
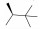
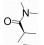
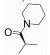
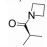
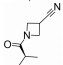
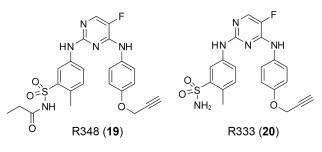
R348(19)是Rigel公司研发的一种选择性JAK-3/SYK (spleen tyrosine kinase)抑制剂[54], R348是R333 (20)分子的前药, 在胃肠道和肝脏内, R348分子在酯酶的作用下, 脱去丙酰基代谢为活性代谢物R333发挥作用, R333可以选择性抑制JAK1/3。实验研究发现, R348代谢为R333后, 半衰期延长, 药代动力学性质更好。2013年10月, R333治疗慢性皮肤疾病盘状红斑狼疮的一项Ⅱ期临床试验失败, 2014年8月, 一项测定R348用于干眼症治疗的Ⅱ期临床因未达到预期测试目标而终止, 但R348用于移植物抗宿主病(graft versus host disease, GVHD)患者干眼症治疗的Ⅱ期临床试验仍在进行[55, 56]。
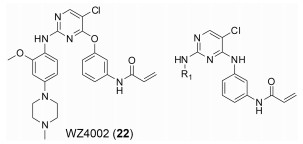
2015年, Tan等[57]报道了一系列具有高选择性JAK-3抑制活性的2, 4-二取代嘧啶类化合物, 其中化合物21抑制JAK-3活性小于0.5 nmol·L-1, 与tofacitinib相比活性更高, 其在转染的Ba/F3细胞实验中, 对于JAK-1、JAK-2和TYK-2的选择性分别达到110、210和100倍, 其选择性也明显优于tofacitinib。该系列化合物以EGFRT790M抑制剂WZ4002 (22)为先导化合物, 将其结构中的醚键用氨基亚甲基键取代后, 活性得到显著提升(23), 分析化合物23与JAK-3的复合物发现(图 5), 化合物苯胺基嘧啶部分与Leu905形成了两个氢键, 丙烯酰胺部分与Cys909形成了共价键, 该共价键与化合物高JAK-3选择性密切相关。以化合物23为先导化合物继续优化, R1吡嗪基被吗啉基取代后, 尽管没有苯环上的2-甲氧基, 化合物24同样能有效抑制JAK-3。R1用1-甲基吡唑基取代(25), 活性进一步提升, 而且选择性和小鼠肝微粒体半衰期(mouse liver microsomal half-life, MLM t1/2)值均保持稳定。在此基础上, 用甲基乙基醚取代甲基(26), 化合物活性进一步提升, 并且表现出了对JAK-3转染的Ba/F3细胞系超出JAK-1和JAK-2等转染的Ba/F3细胞系390倍的选择性, 然而却显示出相对较差的MLM t1/2值。进一步对化合物26进行修饰, 用羟基取代甲基醚基(21), 化合物活性未表现出明显变化, 对JAK-3转染的Ba/F3细胞系的选择性有一定程度降低, 但其MLM t1/2值是化合物26的3倍。该系列化合物具有优异的细胞活性和选择性, 以期能尽快进入临床阶段(表 4)。

|
Figure 5 Binding mode of compound 23 within the JAK-3 binding site[57] |
| Table 4 SAR studies for 2, 4-substituted pyrimidine analogues. aTEL: TEL gene, also known as ETV-6 gene |
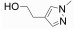

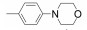
|
NIBR3049(27)是一类对苯基吲哚马来酰亚胺类化合物, 由Novartis公司研发筛选得到[58]。NIBR3049对JAK-1、JAK-2、JAK-3和TYK-2的IC50分别为1 017.0 nmol·L-1、2 550.0 nmol·L-1、8.0 nmol·L-1和8 055.0 nmol·L-1, 与tofacitinib相比, NIBR3049体外激酶实验对JAK-3的选择性优于tofacitinib, 而在细胞实验中, NIBR3049对JAK-3的选择性要低于tofacitinib。NIBR3049的渗透性和细胞浓度与tofacitinib较为接近。化合物28对JAK-3的IC50为27.0 nmol·L-1, 其对JAK-3的选择性分别是对JAK-1、JAK-2和TYK-2的9、10和160倍, 同时具有成为一个先导化合物的潜质, 化合物28与JAK-3复合物表明(图 6), 马来酰亚胺环上的亚氨基和羰基分别与Glu903和Leu905形成两个氢键, 并且化合物28与Ala853和Leu956两个氨基酸的残基形成疏水作用, 马来酰亚胺环的另一个羰基氧通过水桥与氨基酸残基Asp967形成氢键作用, 而在JAK-1和JAK-2的活性腔中, 由于马来酰亚胺的构象不能形成水分子介导的水桥作用。以化合物28为先导化合物继续优化, R1被吗啉(29)或哌嗪(30)取代, 抑制活性会降低, 但选择性部分升高。当哌嗪中的亚胺被醛基取代后, 得到进一步改造的化合物31, 该化合物的活性和选择性均得到了提高。继续优化, 用2-羟基-2-甲基丙酰基取代醛基(27), 化合物活性和选择性进一步提升, 得到最终产物NIBR3049 (表 5)。

|
Figure 6 Detailed view of the X-ray crystal structure of compound 28 (carbons in orange) bound to the ATP-binding site of JAK-3[58] |
| Table 5 SAR studies for phenyl-indolyl maleimide analogues |
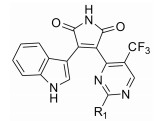
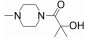
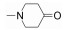
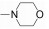
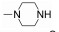
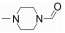
除上述选择性JAK-3抑制剂结构外, 其他对JAK-3有抑制活性的化合物还有很多, 其中有DNX-04042和ABT-494等, 这些药物都在临床试验阶段。
DNX-04042是Dynamix公司研发的选择性JAK-3抑制剂, 其对JAK-2/JAK-3选择性大于1 500倍, 在大鼠胶原诱导的关节炎(collagen induced arthritis, CIA)实验中, 发现其治疗效果优于tofacitinib。同时临床前最大耐受量(maximal tolerance dose, MTD)实验显示其具有良好的药理作用和优良的安全性[4]。2013年11月, 该公司宣布与CleveXel合作研发该化合物, 用于治疗RA, 更多信息尚未公开。
ABT-494是AbbVie公司开发的一种非ATP竞争性JAK抑制剂, 也被称为第二代JAK激酶抑制剂, 目前该化合物的结构尚未公开。该药物通过与ATP结合位点外的其他区域相互作用提高了对JAK-1的选择性, 在细胞特异性评价中对JAK2/JAK1选择性高达74倍。在大鼠AIA模型中, 与tofacitinib相比, ABT-494不影响EPO信号通路和外周NK细胞数目。该结果与14天的Ⅰ期临床研究结果具有一致性[59]。2013年10月, AbbVie启动ABT-494为期12周的Ⅱ期临床试验, 该研究用于抗TNF治疗无效的RA患者, 其除每日2次3 mg在ACR20缓解方面未见显著差异外, 每日2次6、12 mg和每日1次24 mg均达到ACR20缓解率终点[60]。2015年9月, AbbVie出于剂量和安全性考虑决定放弃filgotinib转而开发ABT-494, 并于2016年1月8日启动了ABT-494的Ⅲ期临床研究。ABT-494的研发为非ATP竞争性JAK-3抑制剂的研发提供了思路和借鉴, 因而在本文中进行了特别介绍, 以期非ATP竞争性JAK-3抑制剂能尽早研发出来, 为尚未满足临床治疗需求的RA患者提供新的解决方案。
4 结语与展望JAK-STAT信号通路在细胞分化及增殖过程中起到了重要的作用, JAK活性的改变也将导致该通路信号传递的改变, 进而影响细胞功能。基于JAK激酶家族在JAK-STAT信号传递中的关键作用, 以及JAK-3激酶特定的组织细胞分布, 使得JAK-3成为类风湿性关节炎等疾病良好的治疗靶点。到目前为止, tofacitinib经FDA批准已应用于RA的治疗, 同时还有多个小分子化合物进入临床研究阶段。
目前JAK-3抑制剂主要用于中重度RA患者的治疗, 该类药物在RA的治疗中表现出良好的治疗效果和较好的安全性, 但其长期安全性仍有待进一步研究。上市药物tofacitinib临床研究过程中发现, 使用该药物后会导致一定的不良反应, 包括感染、结核、肿瘤和肝损伤等, 而上述部分不良反应被认为可能与tofacitinib对JAK-3选择性不足相关。欧洲药品管理局人用药品委员会(CHMP)综合考虑该药物对RA治疗的有效性及其不良反应[4, 61], 未批准该药物在欧洲上市。所以提高JAK-3抑制剂的选择性、减少毒副作用是该研究领域亟待解决的关键问题。
JAK激酶家族4个亚型的ATP结合位点同源性较高, 结构差异性较小, 这是导致部分JAK-3抑制剂选择性不高的重要原因, 所以开发有针对性的选择性抑制剂并不容易。解决这一问题或许可以从两方面入手:首先, 目前研究的JAK激酶抑制剂多为ATP竞争性抑制剂, 其主要通过氢键等作用, 竞争性抑制ATP与JAK激酶的结合。虽然不同JAK激酶亚型的ATP结合位点差异性很小, 但仍可以利用各激酶结构域的微小差异来提高JAK-3抑制剂的选择性, 例如: JAK-3激酶蛋白909位氨基酸残基为半胱氨酸(而其他JAK亚型对应位置为丝氨酸), 若JAK-3抑制剂与该氨基酸残基形成共价作用或许将取得较好的结果, 而且相关研究已经在开展中[57]。其次, 由于JAK激酶存在同源性高和差异性小的问题, 因而, 非ATP竞争性抑制剂也将成为选择性JAK-3抑制剂研究的热门领域。
相信随着细胞分子生物学研究的不断深入和筛选手段的快速发展, 选择性JAK-3抑制剂的研究将取得重大进展, 届时也将为类风湿性关节炎、牛皮癣、炎性肠炎、器官移植和肿瘤等疾病的治疗提供新途径。
| [1] | Darnell JE Jr, Kerr IM, Stark GR. JAK-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins[J]. Science , 1994, 264 :1415–1421. DOI:10.1126/science.8197455 |
| [2] | O'Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease[J]. Immunity , 2012, 36 :542–550. DOI:10.1016/j.immuni.2012.03.014 |
| [3] | Smirnova OV, Ostroukhova TY, Bogorad RL. JAK-STAT pathway in carcinogenesis:is it relevant to cholangiocarcinoma progression?[J]. World J Gastroenterol , 2007, 13 :6478–6491. DOI:10.3748/wjg.13.6478 |
| [4] | Norman P. Selective JAK inhibitors in development for rheumatoid arthritis[J]. Expert Opin Investig Drugs , 2014, 23 :1067–1077. DOI:10.1517/13543784.2014.918604 |
| [5] | Sohn SJ, Forbush KA, Nguyen N, et al. Requirement for JAK3 in mature T cells:its role in regulation of T cell homeostasis[J]. J Immunol , 1998, 160 :2130–2138. |
| [6] | Wu W, Sun XH. Janus kinase 3:the controller and the controlled[J]. Acta Biochim Biophys Sin , 2011, 44 :187–196. |
| [7] | Saharinen P, Silvennoinen O. The pseudokinase domain is required for suppression of basal activity of JAK2 and JAK3 tyrosine kinases and for cytokine-inducible activation of signal transduction[J]. J Biol Chem , 2002, 277 :47954–47963. DOI:10.1074/jbc.M205156200 |
| [8] | Chen M, Cheng A, Candotti F, et al. Complex effects of naturally occurring mutations in the JAK3 pseudokinase domain:evidence for interactions between the kinase and pseudokinase domains[J]. Mol Cell Biol , 2000, 20 :947–956. DOI:10.1128/MCB.20.3.947-956.2000 |
| [9] | Ghoreschi K, Gadina M. JAKpot! New small molecules in autoimmune and inflammatory diseases[J]. Exp Dermatol , 2014, 23 :7–11. |
| [10] | Wrobleski ST, Pitts WJ. Advances in the discovery of small molecule JAK3 inhibitors[J]. Annu Rep Med Chem , 2009, 44 :247–264. DOI:10.1016/S0065-7743(09)04412-1 |
| [11] | Lai SY, Johnson FM. Defining the role of the JAK-STAT pathway in head and neck and thoracic malignancies:implications for future therapeutic approaches[J]. Drug Resist Updat , 2010, 13 :67–78. DOI:10.1016/j.drup.2010.04.001 |
| [12] | Cornejo MG, Kharas MG, Werneck MB, et al. Constitutive JAK3 activation induces lymphoproliferative syndromes in murine bone marrow transplantation models[J]. Blood , 2009, 113 :2746–2754. DOI:10.1182/blood-2008-06-164368 |
| [13] | Candotti F, Oakes SA, Johnston JA, et al. Structural and fu nctional basis for JAK3-deficient severe combined immunodeficiency[J]. Blood , 1997, 90 :3996–4003. |
| [14] | O'Sullivan LA, Liongue C, Lewis RS, et al. Cytokine receptor signaling through the JAK-Sta-Socs pathway in disease[J]. Mol Immunol , 2007, 44 :2497–2506. DOI:10.1016/j.molimm.2006.11.025 |
| [15] | Huang M, Rong Y, Ning HX, et al. Establishment of highthroughput drug screening cell models based on JAK-STAT signal pathway[J]. Acta Pharm Sin (药学学报) , 2004, 39 :164–167. |
| [16] | Ivashkiv LB, Hu X. Signaling by STATs[J]. Arthritis Res Ther , 2004, 6 :159–168. DOI:10.1186/ar1197 |
| [17] | Menet CJ, Rompaey LV, Geney R. Advances in the discovery of selective JAK inhibitors[J]. Prog Med Chem , 2013, 52 :153–223. DOI:10.1016/B978-0-444-62652-3.00004-1 |
| [18] | Pesu M, Laurence A, Kishore N, et al. Therapeutic targeting of Janus kinases[J]. Immunol Rev , 2008, 223 :132–142. DOI:10.1111/imr.2008.223.issue-1 |
| [19] | Vainchenker W, Dusa A, Constantinescu SN. JAKs in pathology:role of Janus kinases in hematopoietic malignancies and immunodeficiencies[J]. Semin Cell Dev Biol , 2008, 19 :385–393. DOI:10.1016/j.semcdb.2008.07.002 |
| [20] | Ghoreschi K, Laurence A, O'Shea JJ. Selectivity and therapeutic inhibition of kinases:to be or not to be?[J]. Nat Immunol , 2009, 10 :356–360. DOI:10.1038/ni.1701 |
| [21] | Villa A, Sironi M, Macchi P, et al. Monocyte function in a severe combined immunodeficient patient with a donor splice site mutation in the JAK3 gene[J]. Blood , 1996, 88 :817–823. |
| [22] | Yang QJ, Yu JD, Pan DS, et al. Advance in research of selective JAK inhibitors for treating rheumatoid arthritis[J]. Chin J New Drug (中国新药杂志) , 2015, 24 :39–45. |
| [23] | Riese RJ, Krishnaswami S, Kremer J. Inhibition of JAK kinases in patients with rheumatoid arthritis:scientific rationale and clinical outcomes[J]. Best Pract Res Clin Rh eumatol , 2010, 24 :513–526. DOI:10.1016/j.berh.2010.02.003 |
| [24] | Yamaoka K, Saharinen P, Pesu M, et al. The Janus kinases (JAKs)[J]. Genome Biol , 2004, 5 :253. DOI:10.1186/gb-2004-5-12-253 |
| [25] | Yamaoka K, Min B, Zhou YJ, et al. JAK3 negatively regulates dendritic-cell cytokine production and survival[J]. Blood , 2005, 106 :3227–3233. DOI:10.1182/blood-2005-02-0769 |
| [26] | Kisseleva T, Bhattacharya S, Braunstein J, et al. Signaling through the JAK/STAT pathway, recent advances and future challenges[J]. Gene , 2002, 285 :1–24. DOI:10.1016/S0378-1119(02)00398-0 |
| [27] | MacFarlane LA, Todd DJ. Kinase inhibitors:the next generation of therapies in the treatment of rheumatoid arthritis[J]. In J Rheum Dis , 2014, 17 :359–368. DOI:10.1111/apl.2014.17.issue-4 |
| [28] | Walker JG, Ahern MJ, Coleman M, et al. Changes in synovial tissue JAK-STAT expression in rheumatoid arthritis in response to successful DMARD treatment[J]. Ann Rheum Dis , 2006, 65 :1558–1564. DOI:10.1136/ard.2005.050385 |
| [29] | Burmester GR, Feist E, Dörner T. Emerging cell and cytokine targets in rheumatoid arthritis[J]. Nat Rev Rh eumatol , 2014, 10 :77–88. |
| [30] | Ivashkiv LB, Hu X. The JAK/STAT pathway in rheumatoid arthritis:pathogenic or protective?[J]. Arthritis Rheum , 2003, 48 :2092–2096. DOI:10.1002/(ISSN)1529-0131 |
| [31] | Wang F, Sengupta TK, Zhong Z, et al. Regulation of the balance of cytokine production and the signal transducer and activator of transcription (STAT) transcription factor activity by cytokines and inflammatory synovial fluids[J]. J Exp Med , 1995, 182 :1825–1831. DOI:10.1084/jem.182.6.1825 |
| [32] | Krause A, Scaletta N, Ji JD, et al. Rheumatoid arthritis synoviocyte survival is dependent on Stat3[J]. J Immunol , 2002, 169 :6610–6616. DOI:10.4049/jimmunol.169.11.6610 |
| [33] | de Hooge ASK, Van de Loo FAJ, Koenders MI, et al. Local activation of STAT-1 and STAT-3 in the inflamed synovium during zymosan-induced arthritis:exacerbation of joint inflammation in STAT-1 gene-knockout mice[J]. Arthritis Rh eum , 2004, 50 :2014–2023. DOI:10.1002/(ISSN)1529-0131 |
| [34] | Kasperkovitz PV, Verbeet NL, Smeets TJ, et al. Activation of the STAT1 pathway in rheumatoid arthritis[J]. Ann Rheum Dis , 2004, 63 :233–239. DOI:10.1136/ard.2003.013276 |
| [35] | Finnegan A, Grusby MJ, Kaplan CD, et al. IL-4 and IL-12 regulate proteoglycan-induced arthritis through Stat-dependent mechanisms[J]. J Immunol , 2002, 169 :3345–3352. DOI:10.4049/jimmunol.169.6.3345 |
| [36] | Boggon TJ, Li Y, Manley PW, et al. Crystal structure of the JAK3 kinase domain in complex with a staurosporine analog[J]. Blood , 2005, 106 :996–1002. DOI:10.1182/blood-2005-02-0707 |
| [37] | Vassilev AO, Tibbles HE, DuMez D, et al. Targeting JAK3 and BTK tyrosine kinases with rationally-designed inhibitors[J]. Curr Drug Targets , 2006, 7 :327–343. DOI:10.2174/138945006776054997 |
| [38] | Flanagan ME, Blumenkopf TA, Brissette WH, et al. Discovery of CP-690, 550:a potent and selective Janus kinase (JAK) inhibitor for the treatment of autoimmune diseases and organ transplant rejection[J]. J Med Chem , 2010, 53 :8468–8484. DOI:10.1021/jm1004286 |
| [39] | Chrencik JE, Patny A, Leung IK, et al. Structural and thermodynamic characterization of the TYK2 and JAK3 kinase domains in complex with CP-690550 and CMP-6[J]. J Mol Biol , 2010, 400 :413–433. DOI:10.1016/j.jmb.2010.05.020 |
| [40] | Meyer DM, Jesson MI, Li X, et al. Anti-inflammatory activity and neutrophil reductions mediated by the JAK1/JAK3 inhibitor, CP-690, 550, in rat adjuvant-induced arthritis[J]. J Inflamm , 2010, 7 :41. DOI:10.1186/1476-9255-7-41 |
| [41] | van der Heijde D, Tanaka Y, Fleischmann R, et al. Tofacitinib (CP-690, 550) in patients with rheumatoid arthritis receiving methotrexate:twelve-month data from a twenty-four-month phase Ⅲ randomized radiographic study[J]. Arthritis Rheum , 2013, 65 :559–570. DOI:10.1002/art.37816 |
| [42] | Burmester G, Blanco R, Charles-Schoeman C, et al. Tofacitinib (CP-690, 550), an oral Janus kinase inhibitor, in combination with methotrexate, in patients with active rheumatoid arthritis with an inadequate response to tumor necrosis factor-inhibitors:a 6-month phase 3 study[J]. Arthritis Rheum , 2011, 63 :S279. |
| [43] | Kulagowski JJ, Blair W, Bull RJ, et al. Identification of imidazo-pyrrolopyridines as novel and potent JAK1 inhibitors[J]. J Med Chem , 2012, 55 :5901–5921. DOI:10.1021/jm300438j |
| [44] | Vincenti F, Tedesco Silva H, Busque S, et al. Randomized phase 2b trial of tofacitinib (CP-690, 550) in de novo kidney transplant patients:efficacy, renal function and safety at 1 year[J]. Am J Transplant , 2012, 12 :2446–2456. DOI:10.1111/ajt.2012.12.issue-9 |
| [45] | Boy MG, Wang C, Wilkinson BE, et al. Double-blind, placebo-controlled, dose-escalation study to evaluate the pharmacologic effect of CP-690, 550 in patients with psoriasis[J]. J Invest Dermatol , 2009, 129 :2299–2302. DOI:10.1038/jid.2009.25 |
| [46] | Sandborn WJ, Ghosh S, Panes J, et al. Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis[J]. N Engl J Med , 2012, 367 :616–624. DOI:10.1056/NEJMoa1112168 |
| [47] | Yamazaki S, Inami M, Ito M, et al. ASP015K:a novel JAK inhibitor demonstrated potent efficacy in adjuvant-induced arthritis model in rats[J]. Arthritis Rheum , 2012, 64 :2084. |
| [48] | Takeuchi T, Tanaka Y, Iwasaki M, et al. OP0149 A phase 2B study of an oral JAK inhibitor ASP015K monotherapy in Japanese patients with moderate to severe rheumatoid arthritis[J]. Ann Rheum Dis , 2014, 73 :117. |
| [49] | Janssen announces worldwide agreement to develop and commercialize JAK inhibitor for immunological diseases[EB/OL]. PR Newswire:Janssen Biotech Inc, 2012[2012-10-01]. http://finance.boston.com/boston/news/read?GUID=22390729. |
| [50] | Astellas up on pipeline update, oncology in focus-analyst blog[EB/OL]. Zacks Equity Research, 2014[2014-07-16]. http://www.nasdaq.com/article/astellas-up-on-pipeline-update-oncologyin-focus-analyst-blog-cm370730. |
| [51] | Genovese MC, van Vollenhoven R, Bloom BJ, et al. A phase 2b, 12-week study of VX-509, an oral selective Janus kinase 3 inhibitor, in combination with background methotrexate in rheumatoid arthritis[J]. Arthritis Rheum , 2013, 65 :3320. |
| [52] | Hoock T, Hogan J, Mahajan S, et al. VX-509, an orally available janus kinase 3(JAK3) specific inhibitor, showed robust activity in pre-clinical models of aberrant immune/inflammatory function[J/OL]. Arthritis Rheum, 2011, 63:1136. http://www.blackwellpublishing.com/acrmeeting/abstract.asp?MeetingID=781&id=95876. |
| [53] | Soth M, Hermann JC, Yee C, et al. 3-Amido pyrrolopyrazine JAK kinase inhibitors:development of a JAK3 vs JAK1 selective inhibitor and evaluation in cellular and in vivo models[J]. J Med Chem , 2013, 56 :345–356. DOI:10.1021/jm301646k |
| [54] | Deuse T, Velotta JB, Hoyt G, et al. Novel immunosuppression:R348, a JAK3-and Syk-inhibitor attenuates acute cardiac allograft rejection[J]. Transplantation , 2008, 85 :885–892. DOI:10.1097/TP.0b013e318166acc4 |
| [55] | Chang BY, Zhao F, He X, et al. JAK3 inhibition significantly attenuates psoriasiform skin inflammation in CD18 mutant PL/J mice[J]. J Immunol , 2009, 183 :2183–2192. DOI:10.4049/jimmunol.0804063 |
| [56] | Velotta JB, Deuse T, Haddad M, et al. A novel JAK3 inhibitor, R348, attenuates chronic airway allograft rejection[J]. Transplantation , 2009, 87 :653–659. DOI:10.1097/TP.0b013e318196110f |
| [57] | Tan L, Akahane K, McNally R, et al. Development of selective covalent Janus kinase 3 inhibitors[J]. J Med Chem , 2015, 58 :6589–6606. DOI:10.1021/acs.jmedchem.5b00710 |
| [58] | Thoma G, Nuninger F, Falchetto R, et al. Identification of a potent Janus kinase 3 inhibitor with high selectivity within the Janus kinase family[J]. J Med Chem , 2010, 54 :284–288. |
| [59] | Voss J, Graff C, Schwartz A, et al. Pharmacodynamics of a novel JAK1 selective inhibitor in rat arthritis and anemia models and in healthy human subjects[J/OL]. Arthritis Rh eum, 2013, 65:2374. http://www.blackwellpublishing.com/acrmeeting/abstract.asp?MeetingID=799&id=109741. |
| [60] | Voss J, Graff C, Schwartz A, et al. THU0127 Pharmacodynamics of a novel JAK1 selective inhibitor in rat arthritis and anemia models and in healthy human subjects[J]. Ann Rh eum Dis , 2014, 73 :222. DOI:10.1136/annrheumdis-2014-eular.6179 |
| [61] | van Vollenhoven RF, Fleischmann R, Cohen S, et al. To facitinib or adalimumab versus placebo in rheumatoid arthritis[J]. N Engl J Med , 2012, 367 :508–519. DOI:10.1056/NEJMoa1112072 |