 2016, Vol. 51
2016, Vol. 51

HIV病毒侵入宿主并复制增殖,有3个重要的酶参与: 逆转录酶、整合酶和蛋白酶。抑制其中任何一个酶都可以阻止病毒的繁殖,成为治疗艾滋病的药物。本文叙述作用于整合酶的首创性药物雷特格韦的化学结构的构建历程。
1 HIV病毒整合酶的作用机制HIV病毒进入宿主细胞后,首先在逆转录酶的催化下,将自带的两条RNA链逆转录为两条互补的DNA链,然后由整合酶发生作用。整合酶的作用是使病毒DNA与宿主细胞的DNA分子经缩合而连接成为一体,为下一步的转录和翻译 (即复制病毒自己) 做好准备。整合过程有镁离子作为辅酶在催化中心参与反应。
整合过程包括3个步骤: ① 3' 的加工,是整合酶识别细胞浆内新合成的HIV双链DNA末端四碱基CAGT,结合生成整合前复合物,然后在3' 末端切下两个核苷酸,露出高度保守的CA末端,使病毒DNA 3' 末端的羟基暴露出来用于结合宿主DNA; ② 链转移: 加工形成的病毒DNA蛋白复合物进入细胞核内,整合酶在宿主DNA上切出间隔5个碱基的交错切口,病毒DNA的3' 端带有游离羟基的碱基与宿主DNA的5' 端共价连接起来; ③ 修复阶段: 首先将病毒DNA 5' 端多出的两个未配对的碱基去除,再将缺口连接上,经宿主细胞的酶修补病毒DNA与宿主DNA之间的裂隙并结合在一起,完成了整合过程。
在3' 的加工和链转移中不仅需要二价镁离子参与,而且Mg2+ 也在整合酶与病毒DNA结合中起组 装作用。研究整合酶抑制剂可以从阻断Mg2+ 履行上述结合功能入手。在核酶或多聚核苷酸转移酶等超 家族中,整合酶的催化中心以及与Mg2+ 螯合的氨基酸残基是相当保守和固定的,例如HIV整合酶和丙肝B病毒NS5b依赖于RNA聚合酶的催化中心是 相似的。
2 苗头化合物来自于丙肝病毒的RNA聚合酶抑制剂 2.1 由二酮酸到类药的二羟基嘧啶甲酸默克公司研制HIV整合酶抑制剂来自于对丙肝病毒的RNA 聚合酶的化合物,有抑制活性的苯基-1,3-二酮丙酸 (1 ) 对HCV聚合酶的IC50 = 5.7 μmol·L-1 (Summa V,Petrocchi A,Pace P,et al. Discovery of alpha,gamma- diketo acids as potent selective and reversible inhibitors of hepatitis C virus NS5b RNA-dependent RNA polymerase. J Med Chem,2004,47: 14-17),另一个袂康酸衍生物 (2 ,meconic acid) 对聚合酶的IC50 = 2.25 μmol·L-1 (Pace P,Nizi E,Pacini B,et al. The monoethyl ester of meconic acid is an active site inhibitor of HCV NS5B RNA-dependent RNA polymerase. Bioorg Med Chem Lett,2004,14,3257-3261),这两个化合物都具有可与二价金属离子螯合的基团: 羰基、羧基和酸性羟基,而且它们对聚合酶可发生竞争性抑制,提示结合的位点是相同的。
从化学结构上看,1 和2 不像药物分子,与成药性相距甚远,前者可与氨基酸发生不可逆的共价键结合,后者化学性质不稳定 (容易脱羧)。通过研究羰基与羧基 (或羟基) 的空间距离,移植在类药的骨架上,设计合成的化合物中发现3 对HCV聚合酶有初步活性,IC50 = 30 μmol·L-1。在苯环上作不同取代,发现3-羟基化合物 (4 ) 的活性显著提高,IC50 = 5.8 μmol·L-1,进而将N1甲基化,使C6固定为酮式结构 (5 ) (这个操作在后面优化中反复应用),IC50 = 6.0 μmol·L-1。化合物3 、4 和5 具有化学稳定性,然而对HIV整合酶没有抑制活性 (Summa V,Petrocchi A,Matassa VG,et al. HCV NS5b RNA-dependent RNA polymerase inhibitors: from α,γ-diketoacids to 4,5-dihydroxypyrimidine- or 3-methyl-5-hydroxypyrimidinone carboxylic acids. Design and synthesis. J Med Chem,2004,47: 5336-5339)。
2.2 嘧啶酰胺显示对整合酶的活性为了获得对HIV整合酶有抑制作用的结构类型,变换C2连接的芳环和对4位羧基进行衍生化,发现在一系列芳烷胺的化合物中,6 具有较高的抑制整合酶活性 (测定链转移抑制作用),IC50 = 85 nmol·L-1,虽然生物利用度较低 (F = 15%),但清除率很小 (CLp = 5 mL·min-1·kg-1)。更重要的是6 对HCV聚合酶没有抑制作用,因而定为研发整合酶抑制剂的先导化合物。
2.3 苄基的优化对苄基进行了广泛的结构优化,合成了200多个酰胺化合物,构效关系表明,苄胺的-CH2- 非常重要,因为苯胺的活性很低 (IC50 = 1 μmol·L-1),苯环若被极性较大的芳环置换则活性 下降,提示苄基与整合酶的结合位点处于非极性环境。固定为苯环后,对环上不同位置作各种基团取 代,发现4-F化合物 (7 ) 的活性显著提高,IC50 = 10 nmol·L-1,且对正常细胞没有细胞毒作用,大鼠药代动力学实验表明,口服生利用度F = 29%,有较低的血浆清除率 (CLp = 11 mL·min-1·kg-1),然而化合物7 对感染HIV细胞的抑制活性很弱。不过可认为7 是个里程碑式的化合物 (Petrocchi A,Koch U,Matassa VG,et al. From dihydroxypyrimidine carboxylic acids to carboxamide HIV-1 integrase inhibitors: SAR around the amide moiety. Bioorg Med Chem Lett,2007,17: 350-353)。
2.4 改善过膜和物化性质—嘧啶环2 位的优化, 引入叔氮原子在嘧啶环4位优化为p-氟苄酰胺侧链后,下一步是变换2位取代基以提高对感染细胞的活性。构效关系表明,活性对2位的变换是不敏感的: 去除噻吩环 (2位无取代),或2-甲基、2-异丙基、2-甲苯基、2-苄基或2-吡啶基化合物对抑制活性影响较小 (IC50 = 20~60 nmol·L-1),推论在这个位置连接的基团或片段较少与酶接触,不影响酶活性,因而可在2位作较大范围的结构变换,借助优化物理化学性质以提高过膜性,降低与血浆蛋白的结合率(例如7 与人血浆蛋白的结合率 > 99%,进入细胞中的游离药物分子达不到有效浓度),提高抑制感染细胞的活性。
以2-苄基化合物为母体,引入碱性氮原子 (叔胺),明显提高了过膜能力,从而缩小了抑制酶和抑制感染细胞的活性差距,在众多化合物中,8 抑制整合酶链转移的活性IC50 = 0.2 μmol·L-1,抑制含有10%胎牛血清 (10% FBS) 的感染HIV细胞的IC95 = 0.31 μmol·L-1 (反映穿细胞膜和抑酶能力)。8 的大鼠和犬的口服生物利用度分别为F大鼠 = 59% 和F犬 = 93%; 犬的血浆清除率CLp = 0.5 mL·min-1·kg-1,血浆半衰期t1/2 = 6.0 h,对细胞色素P450未见抑制作用。

|
然而用含有50%正常人血清的感染细胞 (50% NHS,反映化合物的体内活性) 评价化合物8 的活性却很弱,推测是因为有较高的logP值,与血浆蛋白 亲和力高而降低了活性。为此,去除苯环,保留碱性氮原子。
2.5 2位含有碱性氮的饱和环的变换去除化合物8 的2位苯环,环合成哌啶,哌啶连接于2、3或4位,发现2-哌啶基化合物9 的活性 (IC50 = 0.13 μmol·L-1) 强于3-或4-哌啶基化合物,但对感染细胞 (10% FBS IC95 = 1.46 μmol·L-1) 的活性和体内活性 (50% NHS IC95 = 5.83 μmol·L-1) 较低,推测是增加了N-H使氢键增多,不利于过膜和药代。哌啶被N甲基化如化合物10 的体外活性、细胞活性和预测的体内活性相近,高于化合物9 。N,N-二甲基哌嗪化合物11 ,尤其是N-甲基吗啉化合物12 有更高的活性。
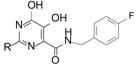

为了进一步提高9 ~12 体内活性,将N1甲基化,使二羟基嘧啶“固定”为酮式结构,合成的有代表性化合物13 ~17 活性强于相应的非N-甲基化合物。
值得一提的是化合物16 ,它的酶抑制活性和在10% FBS存在下的抑制感染细胞的活性低于相应的化合物12 ,但对50% NHS存在下的抑制感染细胞的活性显著增强,尤其是经拆分后的右旋体17 ,3个活性指标均优于12 、16 左旋体和消旋体。17 的盐酸盐有良好的溶解性 (5.8 mg·mL-1) 和适宜的分布系数 (logD = 0.47),犬的口服生物利用度F = 69%,血浆半衰期t1/2 = 7.2 h,低清除率CLp = 2.2 mL·min-1·kg-1,在高剂量下对hERG钾通道和CYP450无抑制作用。然而化合物17 虽然有良好的药效学、药动学和物化性质,但由于有更优良的候选物,未进入临床研究 (Gardelli C,Nizi E,Muraglia E,et al. Discovery and synthesis of HIV integrase inhibitors: development of potent and orally bioavailable N-methyl pyrimidones. J Med Chem,2007,50: 4953-4975)。

与上述平行研发的另一路径是在2位引入含有碱性氮原子的侧链,即去除化合物8 的2位上的苯 环,合成有代表性的2位开链化合物18 ~21 。18 对体外模型具有中等活性。根据上述的哌啶类抑制剂,与C2相连的碳原子以多取代为佳,合成的化合物 19 活性显著提高,进而偕甲基取代化合物 (20 ) 抑制整合酶和阻断感染细胞的活性达到相同的高水平 (Pace P,Di Francesco ME,Gardelli C,et al. Dihydroxypyrimidine-4-carboxamides as novel potent and selective HIV integrase inhibitors. J Med Chem,2007,50: 2225-2239)。


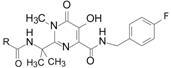
其实,在进行上述优化的同时,还将 N1甲基化的因素导入本系列,以提高酮酸形式的比例。所设计的化合物是固定偕甲基片段,改变胺的结构,而且不拘泥于成盐的碱性,有代表性的化合物如22 ~25 ,其中值得指出的是化合物24 ,该化合物对HIV感染的细胞抑制作用虽然不很强,但是以酰胺的结构出现,为深入优化提供了新的途径,因为可以用不同的羧酸连接。
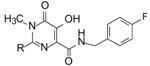
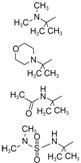
用五元或六元芳杂环替换化合物22 中的甲基,考察对整合酶和感染细胞的抑制活性,有代表性的化合物26 ~30 中,甲基噁二唑化合物30 的细胞活性最高。为确定化合物30 对整合酶的选择性作用,在50 μmol·L-1浓度下对HIV逆转录酶、RNase-H酶和人α,β和γ-聚合酶、HCV聚合酶、重要细胞色素P450以及hERG钾通道等未显示活性; 在50 μmol·L-1浓度下对150种酶 、离子通道和受体也未见活性。
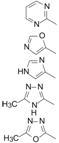
化合物30 制成钾盐 (31 ) 的口服生利用度F大鼠 = 45%,F犬 = 69%~85%; 口服半衰期t1/2(大鼠) = 7.5 h,t1/2(犬) = 7.5 h。犬灌胃2 mg·kg-1和10 mg·kg-1,12 h后血浆药物浓度分别为160和350 nmol·L-1,超过抑制感染细胞的IC95 = 31 nmol·L-1 (含有50% NHS)。对9种整 合酶发生突变的HIV的抑制活性有5株与野生型的IC50值相同,其余4株分别为野生型的IC50值3、6、8和10倍。综合药效活性、药代数据和对突变株的作用,化合物31 比其他待选的化合物优胜,命名为雷特格韦 (31 ,raltegravir) 进入临床研究,经Ⅲ期实验证明是患者感染HIV病毒的治疗药,作为第一个抑制整合酶的口服药物,于2007年经FDA批准在 美国上市 (Summa V,Petrocchi A,Bonelli F,et al. Discovery of raltegravir,a potent,selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. J Med Chem,2008,51: 5843- 5855)。

|