 2016, Vol. 51
2016, Vol. 51



2. 中国科学院上海药物研究所, 上海 201203
2. Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China
盐酸多柔比星 (doxorubicin hydrochloride,DOX·HCl) 是一种广谱蒽环类抗肿瘤药物[1, 2],在生物药剂学分类系统中属BCS III类,水溶性较好,但在小肠上皮细胞的渗透性低 (主要原因是强极性以及P-gp外排作用); 此外,DOX在胃中水解、受CYP450酶降解严重限制了DOX口服给药的生物利用 (低于5%)[3, 4],因此在临床上采取注射给药的方式。目前采取了多种药物载体来提高DOX的口服生物利用度,如中链甘油酯制备的纳米乳[5]、聚合物胶束[6]、脂质纳米粒和聚合物纳米粒等[7]。
脂质纳米粒 (lipid nanoparticles) 或脂质纳米载体(nanostructured lipid carriers,NLCs) 被认为是一种生物相容性好、可有效包载药物和毒性低的给药系统[8]。脂质纳米粒进入消化道后被酶降解释放出脂肪酸对药物具有增溶作用,因而可显著提高难溶性药物的口服生物利用度[9]。纳米粒还能被消化道上皮细胞直接摄取,该吸收机制对强亲水性药物,如多肽蛋白类药物[10, 11]、上皮细胞低渗透性药物[12]的吸收具有一定的优势。然而,胃肠道内复杂的pH、酶环境等对脂质纳米粒具有消化作用[13],导致载体结构被破坏,药物过早大量释放,因而不利于药物的吸收。因此,提高脂质纳米粒在消化道内的稳定性,防止药物过快释放,有助于该载体在BCS III类药物的应用。
本研究希望通过对脂质纳米粒表面进行聚合物电解质层层组装,提高脂质纳米粒在消化道内的稳定性,防止药物过快释放,使脂质纳米粒能有效地被消化道上皮细胞摄取,从而促进DOX的口服吸收。壳聚糖 (chitosan,CS) 是一种生物相容性好可降解的天然高分子多糖,含有大量游离氨基,当pH小于其pKa 6.5时氨基质子化带正电荷。γ-聚谷氨酸 [poly (γ-glutamic acid),γ-PGA] 也是一种生物相容性好的高分子材料,含有丰富的羧基,带有负电荷[14]。利用静电作用使带有相反电荷的聚合物在纳米粒表面层层自组装 (layer-by-layer,LbL) 可形成多层聚合物电解质,使纳米粒形成核−壳结构 [15, 16, 17]。因此,本研究采用层层组装技术制备了多层聚合物电解质组装的脂质纳米粒,并对这几组制剂的理化性质、体外脂消化、细胞摄取以及大鼠体内的口服吸收效果进行了 研究。
材料与方法材料 DOX·HCl (凯里德生物医药技术上海有限公司); 山嵛酸甘油酯 (Compritol 888 ATO,法国Gattefosse公司); 大豆卵磷脂 (Eplkuron 200,德国Degussa公司); 维生素E聚乙二醇1000琥珀酸酯 (TPGS)、普郎尼克F127 (Kutrol) (德国BASF公司); 壳聚糖 (相对分子质量10万,脱乙酰度90%,浙江金壳生物化学有限公司); γ-聚谷氨酸钠 (γ-PGA,相对分子质量100万,上海久谦化工有限公司赠); DMEM培养基 (Sigma公司); 特级胎牛血清 (Hyclone公司); 细胞胰酶消化液、4',6-二脒基-2-苯基吲哚 (DAPI,碧云天生物技术研究所); 三乙醇胺、油酸、胆固醇及其他试剂均购自国药集团化学试剂有限公司; 实验用水为去离子水。
仪器Zetasizer Nano ZS粒度仪 (英国Malvern公司); TAZ4-WS型低速离心机 (上海湘仪离心机有限公司); 探头超声细胞粉碎机SCIENTZ-IID (宁波新芝生物科技股份有限公司); Synergy H1酶标仪 (美国Biotek公司); CO2培养箱HERAcell240 (德国Heraeus公司); BD FACS Calibur型流式细胞仪 (美国Becton Dickinson公司); FV1000激光扫描共聚焦显微镜 (日本Olympus公司); 透射电镜CM-200 (荷兰Philips公司)。
动物SD大鼠,雄性,体重250 ± 20 g,IACUC审批号: 2015-10GY-19。由中国科学院上海药物研究所实验动物中心提供。
载DOX脂质纳米粒的制备采用热熔均质−超声法制备载DOX脂质纳米粒[18]。称取DOX·HCl 5 mg溶于2 mL二氯甲烷/甲醇溶液 (2∶1,v/v) 中,加入三乙醇胺溶液 (DOX·HCl−三乙醇胺1∶3,摩尔比),搅拌过夜,再在氮气流下挥干有机溶剂,得到游离碱DOX固体。称取山嵛酸甘油酯100 mg、油酸10 mg、磷脂10 mg、胆固醇5 mg、TPGS 10 mg与上述DOX (5 mg) 在80 ℃水浴中加热熔融成均一的油相。以Pluronic-F127水溶液 (1%,w/v) 2 mL为水相,并加热至与油相相同温度。在磁力搅拌 (400 r·min−1) 下趁热将水相滴加到油相中,并以13 500 r·min−1高剪切3 min (IKALabortechnik,德国) 得到初乳。将初乳探头超声2 min (功率100 W,超声波发射的脉冲为 2 s,两个脉冲之间停顿2 s) 得到纳米乳。最后将纳米乳在冰浴下搅拌固化1 h,得到载DOX的脂质纳米粒 (DOX-NPs)。
聚合物电解质层层组装脂质纳米粒的制备将上述制备的脂质纳米粒滴加到一定浓度的壳聚糖水溶液中 (用1% 醋酸溶解,调节至pH 5.0),500 r·min−1下搅拌15 min,得到壳聚糖组装的脂质纳米粒 (DOX- NPs/CS); 再将壳聚糖组装的纳米粒溶液滴加入一定浓度的γ-PGA溶液(用蒸馏水溶解,调节至pH 5.0) 中,500 r·min−1下搅拌15 min,即得到两层聚合物电解质组装的纳米粒 (DOX-NPs/CS/γ-PGA)。分别考察了壳聚糖、γ-PGA的浓度对纳米粒zeta电位和粒径的影响。
粒度、粒度分布和zeta电位测定取DOX-NPs、DOX-NPs/CS和DOX-NPs/CS/γ-PGA样品各50 µL,用蒸馏水稀释至1 mL,采用Zetasizer Nano ZS粒度测定仪分别测定样品的平均粒径、粒度分布和zeta电位。
透射电镜观察采用透射电镜 (TEM) 观察DOX-NPs、DOX-NPs/CS和DOX-NPs/CS/γ-PGA的粒子形态。用纯水将样品稀释100倍,取1滴混悬液覆盖在铜网上,滤纸吸取过量的液体。然后在铜网上加1滴2% 磷钨酸染1~2 min,样品在室温下干燥后在透射电子显微镜下观察。
包封率和载药量的测定采用超滤法测定DOX- NPs、DOX-NPs/Cs和DOX-NPs/CS/γ-PGA包封率。取样品200 µL于超滤管中 (MWCO 10 kDa,Sartorius公司),4 500 r·min−1离心10 min,取滤液50 µL置 10 mL量瓶中,用甲醇稀释至刻度,采用酶标仪在 Ex = 480 nm,Em = 590 nm波长下测定溶液中DOX 的荧光强度,得到游离的药物浓度。取样品50 µL置10 mL量瓶中,用甲醇溶解并稀释至刻度; 取溶液于8 000 r·min−1离心10 min,取上清液,用酶标仪测定溶液中DOX的荧光强度,得到纳米粒混悬液中的总药物浓度。取纳米粒混悬液5 mL在4 ℃下20 000 r·min−1离心30 min,除去上层液体,沉淀物冻干,即得纳米粒固体。精密称取适量纳米粒,用甲醇溶解,于8 000 r·min−1离心10 min,取上清液,用酶标仪测定药物浓度,得到纳米粒包载的药物量。采用如下公式计算包封率 (entrapment efficiency,EE) 和载药量 (drug loading,DL)。
| \[{\rm{EE}}\left( \% \right){\rm{ = }}\left( {{C_{\rm{a}}} - {C_{\rm{b}}}} \right)/{C_{\rm{a}}} \times 100\% \] | (1) |
| \[{\rm{DL}}\left( \% \right){\rm{ = }}{W_{\rm{c}}}/{W_{\rm{a}}} \times 100\% \] | (2) |
其中,Ca: 纳米粒悬液中的总药物浓度; Cb: 游离的药物浓度; Wc: 包载的药物量; Wa: 纳米粒的重量。
体外释放度的测定采用透析法测定体外释放特性。取DOX-NPs、DOX-NPs/CS和DOX-NPs/CS/γ-PGA混悬液各1 mL,置透析袋 (截留分子量 = 8 000~14 000 Da) 中。分别以50 mL的pH 1.2盐酸溶液和pH 6.8磷酸盐缓冲液 (PBS) 为释放介质,于37 ℃恒温振荡器中100 r·min−1条件下进行释放度考察,于0.5、1、1.5、2、4、6和8 h取样0.5 mL,并补充相同体积的释放介质,用酶标仪测定药物的荧光强度以定量药物的浓度。
不同pH条件下纳米粒粒径变化及体外脂消化实验研究聚合物电解质组装的纳米粒在模拟胃肠道不同pH环境下的稳定性。分别取DOX-NPs/CS和DOX-NPs/CS/γ-PGA混悬液各0.5 mL置pH 1.2~6.8缓冲液中,37 ℃下孵育30 min,用Zetasizer Nano ZS粒度测定仪测定样品的粒径。
体外脂消化实验[19] 消化介质由2 mmol·L−1 Tris-maleate、150 mmol·L−1 NaCl、5 mmol·L−1 CaCl2、5 mmol·L−1牛胆酸钠、1.25 mmol·L−1磷脂和脂肪酶组成。将消化介质2.5 mL、制剂 (各制剂组1 mL,对照 组: 去离子水) 与适量水混合,至总体积4 mL,37 ℃恒温搅拌,调节pH至6.0,加入脂肪酶溶液1 mL后启动消化反应并计时。以0.1 mol·L−1 NaOH滴定水解反应生成的脂肪酸,使体系pH维持在6.0,在预先设定的时间点记录加入NaOH的体积。
细胞摄取的研究细胞的培养 人结肠癌细胞Caco-2 (来源于中国科学院上海药物研究所) 培养于DMEM培养基 (含10% 胎牛血清和1% 青霉素、链霉素),置于37 ℃、5% CO2 (相对湿度90%) 的培养箱中。每两天换1次,用胰酶/EDTA消化传代。
流式细胞术 将Caco-2细胞以5×104个/孔接种于12孔板,每孔加入DMEM培养基,置于37 ℃、5% CO2的培养箱中培养24 h。待细胞贴壁后,除去原培养液,用PBS清洗细胞3次,加入1 mL用无血清培养基稀释至适当浓度 (DOX,10 µg·mL−1) 的各制剂组: DOX溶液、DOX-NPs、DOX-NPs/CS和DOX-NPs/ CS/γ-PGA组。将细胞置37 ℃、5% CO2的培养箱中孵育2.0 h,弃除含制剂的培养基,用4 ℃ PBS清洗细胞3次。向每孔细胞中加入胰酶消化液200 µL消化3 min,然后加入3倍消化液的含血清培养基终止消化,取出消化后的细胞,置于1.5 mL离心管中,3 000 r·min−1离心2 min。除去上层液,加入PBS 0.5 mL,制成悬浮液,转到离心管,避光保存,流式细胞仪分析。
激光扫描共聚焦显微镜细胞成像 将Caco-2细胞以5×104个/孔接种于12孔板,于37 ℃、5% CO2的培养箱中培养24 h。待细胞贴壁,弃去原培养基,加入无血清的培养基1.0 mL,于37 ℃、5% CO2的培养箱中孵育30 min,用PBS清洗细胞3次,加入1 mL用无血清培养基稀释至适当浓度 (DOX,10 µg·mL−1) 的制剂组,包括DOX溶液、DOX-NPs、DOX-NPs/CS和DOX-NPs/CS/γ-PGA组。将细胞于37 ℃、5% CO2的培养箱中孵育2.0 h。弃去培养液,用PBS清洗细胞3次。用4% 多聚甲醛固定15 min,再用PBS清洗3次。用5 µg·mL−1 DAPI染核,以4 ℃ PBS清洗细胞3次。取出盖玻片,90% 甘油封片后采用激光扫描共聚焦显微镜进行细胞成像。
大鼠口服药动学研究血浆样品处理方法 取血浆样品100 µL置离心管,加甲醇 (含6% HCl) 400 µL,涡旋混合2 min,4 ℃下13 000 r·min−1离心10 min,取上清液200 µL,采用酶标仪测定荧光强度 (Ex = 480 nm,Em = 590 nm)。
DOX血浆样品标准曲线的制备 精密称取DOX适量,用蒸馏水配制浓度为100µg·mL−1储备液。分别取不同体积的DOX溶液,加入空白大鼠血浆,配制成含DOX质量浓度为0.01、0.05、0.1、0.5和1.0 µg·mL−1的大鼠血浆样品,按“血浆样品处理方法”处理样品,测定荧光强度。以空白血浆的荧光值作为本底值。
大鼠口服吸收实验 将24只大鼠随机分为A、B、C和D组,每组6只。A组为DOX对照组,B、C、D组分别为DOX-NPs、DOX-NPs/CS和DOX-NPs/ CS/γ-PGA组。给药前禁食12 h,可自由饮水,每组以10 mg·kg−1 DOX剂量灌胃给药。给药后分别于0、0.5、1、2、4、8和24 h眼眶取血0.3 mL,置肝素处理过的离心管,8 000 r·min−1离心5 min分离血浆,按“血浆样品处理方法”处理样品,测定荧光强度。
统计学方法药动学参数运用DAS2.0分析软件,采用非房室模型分析; 实验数据运用Graph Pad分析,采用t检验比较两组间差异显著性,P < 0.05为具有显著性差异。
结果 1 壳聚糖和γ-PGA用量的筛选制备得到的脂质纳米粒表面呈负电性,利用静电作用可以组装多层聚合物电解质。首先在脂质纳米粒的表面组装带有正电荷的CS,粒子呈正电性,不同浓度的CS溶液在脂质纳米粒表面自组装后,zeta电位和粒径变化如图 1A,B所示,增加CS的浓度,电荷由负电逐渐升高,当质量浓度为0.1 mg·mL−1时,纳米粒的电位开始由负电荷向正电荷转变,此时,粒径明显增大,推测纳米粒之间失去电荷排斥作用,稳定性降低发生聚集。随着CS用量的增加,电位趋向于平衡,粒径较之前降低,继续增加CS的浓度,zeta电位和粒径均变化不大,推测粒子表面吸附CS的量已达到饱和。为控制溶液中游离聚合物电解质的量,选择1.0 mg·mL−1 CS溶液为最佳用量,得到DOX-NPs/ CS。随后,采用γ-PGA进行表面组装,γ-PGA与CS通过静电相互作用吸附在粒子表面。加入不同浓度的γ-PGA后,纳米粒的zeta电位和粒径变化如图 1C,D所示,随着γ-PGA的浓度升高,电位降低,粒径增大; 当γ-PGA的质量浓度为0.5 mg·mL−1时,电位开始发生反转,由正电荷变成电中性,粒径达到最大值,推测由于粒子呈中性,失去粒子之间电荷的排斥作用发生聚集; 继续增加γ-PGA的浓度,zeta电位降低,粒径变小; 再继续增加γ-PGA的浓度,电荷和粒径均变化不大,推测粒子表面吸附的γ-PGA量已达到饱和,因此选择2.5mg·mL−1 γ-PGA溶液为最佳用量,最终得到DOX-NPs/CS/γ-PGA。

|
Figure 1 Effect of CS concentrations on the zeta potential (A) and particle size (B) of layer-by-layer assembled nanoparticles; effect of γ-PGA concentrations on the zeta potential (C) and particle size (D) of layer-by-layer assembled nanoparticles. n = 3,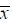 ± s. CS: Chitosan;γ-PGA: Poly (γ-glutamic acid); NPs: Nanoparticles ± s. CS: Chitosan;γ-PGA: Poly (γ-glutamic acid); NPs: Nanoparticles
|
DOX-NPs、DOX-NPs/CS和DOX-NPs/CS/γ-PGA的粒径、粒度分布、电位、包封率和载药量如表 1所示。各组纳米粒的电镜照片如图 2所示,未经聚合物电解质包裹的纳米粒表面光滑、圆整。DOX-NPs/ CS由于表面被聚合物电解质包裹,呈明显的核壳结构; DOX-NPs/CS/γ-PGA表面粗糙,核壳结构更加明显,可能是由于粒子表面组装的聚合物比DOX-NPs/ CS更多,更加致密,因此对内部的脂质成分可能具有更好的保护作用。
|
|
Table 1 Particle size,zeta potential,entrapment efficiency (EE) and drug loading (DL) of DOX-NPs,DOX-NPs/CS,DOX-NPs/CS/ γ-PGA formulations. n = 3,± s. DOX: Doxorubicin; PDI: Polydispersity index
|

|
Figure 2 TEM images of DOX-NPs (A),DOX-NPs/CS (B),DOX-NPs/CS/γ-PGA (C) |
在0.1~2.0 µg·mL−1内,DOX浓度 (C) 对荧光强度 (F) 呈良好的线性关系,其回归方程F = 1 786.3 C + 75.842 (r = 0.999 9,n = 3)。图 3为载DOX各组制剂在模拟胃液 (pH 1.2盐酸溶液) 和模拟肠液 (pH 6.8 PBS) 中的释药曲线。结果显示,在模拟胃液中,DOX-NPs、DOX-NPs/CS和DOX-NPs/CS/γ-PGA组在4 h分别释放约75%、65% 和58% (图 3A); 在模拟肠液中,DOX- NPs、DOX-NPs/CS和DOX-NPs/CS/γ-PGA组在前2 h 释药较快,释放了50% 左右,后6 h释放缓慢,8 h累计释放百分率为60% (图 3B)。

|
Figure 3 Invitro drug release profile of nanoparticles in pH 1.2 HCl solution (A),and in pH 6.8 phosphate buffer solution (PBS,B) at 37 ℃. n = 3,± s
|
DOX-NPs/CS和DOX-NPs/CS/γ-PGA在pH 1.2~6.8下的粒径变化如图 4A,B所示,在不同pH下,DOX-NPs/CS和DOX-NPs/CS/γ-PGA的粒径均无较 大变化,说明在此pH范围内 ,各制剂均较稳定。各制剂的体外脂消化结果如图 4C所示,在1 h内DOX-NPs消耗NaOH的量和速度明显高于DOX-NPs/CS和DOX-NPs/CS/γ-PGA消耗NaOH的量,表明对脂质纳米粒表面进行聚合物电解质层层组装后可延缓脂肪酶对脂质纳米粒的消化作用,有利于纳米粒在消化道内的稳定性,防止药物在消化道内过快释放。

|
Figure 4 Effect of pH value on the particle size stability of DOX-NPs/CS (A) and DOX-NPs/CS/γ-PGA (B); the consumption of NaOH as a function of time for the formulations (C) during lipolysis in vitro |
Caco-2细胞摄取DOX溶液、DOX-NPs、DOX-NPs/CS和DOX-NPs/CS/γ-PGA的激光扫描共聚焦 显微照片如图 5所示,可观察到与DOX-NPs、DOX- NPs/CS、DOX-NPs/CS/γ-PGA组孵育的细胞内荧光 强度明显高于DOX溶液组。流式细胞术定量分析 DOX溶液、DOX-NPs、DOX-NPs/CS和DOX-NPs/ CS/γ-PGA组 在Caco-2细胞中2 h的摄取情况如图 6所示。DOX溶液组在细胞内的摄取量很低,而DOX- NPs、DOX-NPs/CS和DOX-NPs/CS/γ-PGA组的细胞摄取量分别是DOX溶液的3.9、3.8和4.0倍。

|
Figure 5 Laser scanning confocalmicroscope images of cells after incubation of free DOX and DOX formulations for 2 h. Scale bar is 50 µm |

|
Figure 6 Flow cytometry analysis of fluorescence intensity (MFI) of Caco-2 cells after incubation with free DOX,DOX-NPs,DOX-NPs/CS,DOX-NPs/CS/γ-PGA for 2 h. n = 3,± s. **P < 0.01
|
DOX血浆药物浓度测定标准曲线在0.01~1.0 µg·mL−1内线性良好,y = 5 000.4x + 212.68,r = 0.999 3,分析方法的灵敏度和重现性均符合药动学研究的要求。大鼠灌胃给予DOX溶液、DOX-NPs、DOX-NPs/CS和DOX-NPs/CS/γ-PGA后,DOX在大鼠体内的药−时曲线如图 7所示,各药动学参数如表 2所示。DOX溶液组口服吸收极差,Cmax仅为0.18 ± 0.02 µg·mL−1; DOX-NPs、DOX-NPs/CS和DOX-NPs/CS/γ-PGA 3组制剂的Cmax和AUC0−24 h 均显著高于DOX溶液组,其中DOX-NPs/CS/γ-PGA组的吸收AUC0−24 h显著高于DOX-NPs,说明在脂质纳米粒表面组装聚电解质材料可以提高纳米粒在胃肠环境中的稳定性,促进药物的吸收。

|
Figure 7 Plasma concentration-time profiles of DOX in rats after oral administration of different DOX formulations. n = 6,± s
|
|
|
Table 2 Pharmacokinetic parameters of DOX after oral administration of different formulations in rats. n = 6,± s. P < 0.05,**P < 0.01,***P < 0.001 vs DOX-NPs. tmax: Time to maximum plasma concentration; Cmax: Maximum plasma concentration; AUC0−24 h: Area under the curve up to 24 h; F: Relative bioavailability
|
亲脂性药物在脂质纳米粒中的载药量较高,是由于亲脂性药物与脂类材料之间具有较高的相容性。DOX·HCl为极性较强的化合物,与脂质材料的相 容性较弱,因此很难将DOX·HCl载入脂质材料中,包封率低。本文采用油酸与DOX形成离子对,可显著提高DOX在脂质中的包封率。另外将DOX·HCl转化成DOX碱基形式,由于极性的降低可进一步 提高DOX在熔融脂质材料中的溶解度。因此,得到的DOX-NPs、DOX-NPs/CS和DOX-NPs/CS/γ-PGA 3组制剂包封率均可达到95% 以上,载药量在3.5% 左右。
在体外脂消化实验中,DOX-NPs/CS/γ-PGA消耗NaOH的量明显低于DOX-NPs和DOX-NPs/CS消耗NaOH的量,说明组装在脂质纳米粒表面的聚电解质在酶解过程中对纳米粒起到一定的保护作用,多层聚电解质组装的脂质纳米粒被酶解的速率较单层聚电解质组装的脂质纳米粒更慢,原因可能是聚合物电解质在脂质纳米粒和脂肪酶之间形成一定的空间位阻,阻碍脂肪酶作用于脂质纳米粒中的脂质成分的活性位点。
在Caco-2细胞摄取实验中,3组制剂的细胞摄取明显高于DOX溶液组,表明制备成脂质纳米粒后可显著提高细胞对药物的摄取,然而3组纳米粒之间 无显著性差异。表面电荷会影响纳米粒的细胞摄取,纳米粒表面正电性越强越易于被细胞摄取[20]。本实验中,zeta电位的测定是以纯水为分散介质时测定的,
DOX-NPs/CS的zeta电位为17.10 ± 0.36 mV。介质 的pH值、离子强度等对聚合物电解质纳米粒的zeta电位有较大的影响。壳聚糖pKa值为6.5,溶液pH 可影响其链上氨基质子化程度,进而影响纳米粒 表面的zeta电位。细胞摄取实验是在pH 7.4 PBS 无血清培养基中进行的,在此介质中,DOX-NPs、DOX-NPs/CS、DOX-NPs/CS/γ-PGA的zeta电位分别是 −35.6 mV、−5.6 mV和−43.7 mV。因此,在此环境下可能削弱了细胞对DOX-NPs/CS的摄取能力。此外,纳米粒还可能通过一些特殊受体的介导被细胞摄取[21]。本课题组发现含γ-PGA的纳 米粒能通过细胞表面表达的γ-谷氨酰转移酶的介导促进入胞[22]。经这些受体的介导,导致纳米粒能通过一些特殊的摄取机制被细胞摄取,如诱导激发网格蛋白和脂筏介导的内吞作用[23, 24]。另外,表面疏水性也会影响纳米粒的细胞摄取,表面疏水性越强,越易于被细胞摄取,表面未被聚合物电解质修饰的纳米粒可能疏水性比含聚合物电解质的纳米粒强。综上所述,由于多种因素的交叉影响,很难判断何种因素是影响纳米粒入胞的主要因素。
纳米粒经口服给药进入消化道后,消化道内复杂的环境包括pH变化、酶的消化作用对纳米粒均 具有破坏作用,导致纳米粒的粒径增大或被酶降解。DOX-NPs/CS/γ-PGA的口服吸收明显高于DOX-NPs,分析原因是DOX-NPs/CS/γ-PGA能被更稳定地运输至肠上皮细胞,而未组装聚合物电解质的脂质纳米粒DOX/NPs则可能在肠道中被脂肪酶降解,载体结构被破坏,药物泄漏,纳米粒未能运送至肠上皮细胞,导致生物利用度低于DOX-NPs/CS/γ-PGA。
本研究表明,聚合物电解质层层组装的脂质纳米粒可显著提高DOX的口服生物利用度,具有一定的应用前景。对其在胃肠道中的命运和肠上皮细胞对纳米粒的摄取机制方面还有待进一步研究与探索。
结论本研究制备了聚合物电解质层层组装的脂质纳米粒DOX-NPs/CS/γ-PGA,可以有效包载DOX,提高Caco-2细胞对DOX的摄取,采用聚合物电解质层层组装延缓了药物的释放,提高了脂质纳米粒在消化道内的酶解稳定性,并且显著提高了DOX在大鼠体内的口服吸收。结果表明,聚合物电解质包裹的脂质纳米粒对促进低渗透性药物的口服吸收具有一定的意义。
| [1] | Octavia Y, Tocchetti CG, Gabrielson KL, et al. Doxorubicininduced cardiomyopathy:from molecular mechanisms to therapeutic strategies[J]. J Mol Cell Cardiol, 2012, 52:1213-1225. |
| [2] | Primeau AJ, Rendon A, Hedley D, et al. The distribution of the anticancer drug doxorubicin in relation to blood vessels in solid tumors[J]. Clin Cancer Res, 2005, 11:8782-8788. |
| [3] | Kalaria DR, Sharma G, Beniwal V, et al. Design of biodegradable nanoparticles for oral delivery of doxorubicin:in vivo pharmacokinetics and toxicity studies in rats[J]. Pharm Res, 2009, 26:492-501. |
| [4] | Benival DM, Devarajan PV. Lipomer of doxorubicin hydrochloride for enhanced oral bioavailability[J]. Int J Pharm, 2012, 423:554-561. |
| [5] | Kim JE, Yoon IS, Cho HJ, et al. Emulsion-based colloidal nanosystems for oral delivery of doxorubicin:improved intestinal paracellular absorption and alleviated cardiotoxicity[J]. Int J Pharm, 2014, 464:117-126. |
| [6] | Wang J, Li L, Du Y, et al. Improved oral absorption of doxorubicin by amphiphilic copolymer of lysine-linked ditocopherol polyethylene glycol 2000 succinate:in vitro characterization and in vivo evaluation[J]. Mol Pharm, 2015, 12:463-473. |
| [7] | Mei L, Zhang Z, Zhao L, et al. Pharmaceutical nanotechnology for oral delivery of anticancer drugs[J]. Adv Drug Deliv Rev, 2013, 65:880-890. |
| [8] | Mehnert W, Mäder K. Solid lipid nanoparticles:production, characterization and applications[J]. Adv Drug Deliv Rev, 2001, 47:165-196. |
| [9] | Mu H, Holm R, Müllertz A. Lipid-based formulations for oral administration of poorly water-soluble drugs[J]. Int J Pharm, 2013, 453:215-224. |
| [10] | Fan T, Chen C, Guo H, et al. Design and evaluation of solid lipid nanoparticles modified with peptide ligand for oral delivery of protein drugs[J]. Eur J Pharm Biopharm, 2014, 88:518-528. |
| [11] | Almeida AJ, Souto E. Solid lipid nanoparticles as a drug delivery system for peptides and proteins[J]. Adv Drug Deliv Rev, 2007, 59:478-490. |
| [12] | Pawar YB, Purohit H, Valicherla GR, et al. Novel lipid based oral formulation of curcumin:development and optimization by design of experiments approach[J]. Int J Pharm, 2012, 436:617-623. |
| [13] | Porter CJH, Pouton CW, Cuine JF, et al. Enhancing intestinal drug solubilisation using lipid-based delivery systems[J]. Adv Drug Deliv Rev, 2008, 60:673-691. |
| [14] | Sonaje K, Lin YH, Juang JH, et al. In vivo evaluation of safety and efficacy of self-assembled nanoparticles for oral insulin delivery[J]. Biomaterials, 2009, 30:2329-2339. |
| [15] | Shchukina EM, Shchukin DG. Layer-by-layer coated emulsion microparticles as storage and delivery tool[J]. Curr Opin Colloid Interf Sci, 2012, 17:281-289. |
| [16] | Ramasamy T, Tran TH, Choi JY, et al. Layer-by-layer coated lipid-polymer hybrid nanoparticles designed for use in anticancer drug delivery[J]. Carbohydr Polym, 2014, 102:653-661. |
| [17] | Jeon S, Yoo CY, Park SN. Improved stability and skin permeability of sodium hyaluronate-chitosan multilayered liposomes by layer-by-layer electrostatic deposition for quercetin delivery[J]. Colloids Surf B Biointerf, 2015, 129:7-14. |
| [18] | Mussi SV, Silva RC, de Oliveira MC, et al. New approach to improve encapsulation and antitumor activity of doxorubicin loaded in solid lipid nanoparticles[J]. Eur J Pharm Sci, 2013, 48:282-290. |
| [19] | Parmentier J, Thomas N, Müllertz A, et al. Exploring the fate of liposomes in the intestine by dynamic in vitro lipolysis[J]. Int J Pharm, 2012, 437:253-263. |
| [20] | Young JJ, Chen CC, Chen YC, et al. Positively and negatively surface-charged chondroitin sulfate-trimethylchitosan nanoparticles as protein carriers[J]. Carbohydr Polym, 2016, 137:532-540. |
| [21] | Chakraborty A, Jana NR. Clathrin to lipid raft-endocytosis via controlled surface chemistry and efficient perinuclear targeting of nanoparticle[J]. J Phys Chem Lett, 2015, 6:3688-3697. |
| [22] | Zhu Q, Song W, Xia D, et al. A poly-L-glutamic acid functionalized nanocomplex for improved oral drug absorption[J]. J Mater Chem B, 2015, 3:8508-8517. |
| [23] | Chai GH, Hu FQ, Sun J, et al. Transport pathways of solid lipid nanoparticles across madin-darby canine kidney epithelial cell monolayer[J]. Mol Pharm, 2014, 11:3716-3726. |
| [24] | He B, Lin P, Jia Z, et al. The transport mechanisms of polymer nanoparticles in Caco-2 epithelial cells[J]. Biomaterials, 2013, 34:6082-6098. |