 2016, Vol. 51
2016, Vol. 51



离子通道广泛地分布于各种组织细胞的生物膜上,对于细胞膜的电生理具有十分重要的作用。Kv2.1是电压门控钾离子通道中一种重要的亚型,分布于多种组织中,如大脑神经元、胰岛组织等,具有重要的生理药理功能[1, 2]。
缺血性脑卒中发病会导致神经元凋亡,治疗后往往会有失语、偏瘫等后遗症,丧失劳动能力。因此,对于脑卒中患者的神经元保护具有重要意义。研究发现[3],Kv2.1电流是神经元外向延迟整流钾电流的主要成分,脑卒中发病时,钾离子的过度外流和细胞内钾的大量丢失是造成脑缺血细胞损伤和凋亡的主要机制,抑制Kv2.1通道的过度开放则可能发挥对脑缺血细胞损伤的保护作用。
早期发现的Kv2.1选择性阻断剂为一些多肽类化合物,它们来自于蜘蛛、蟾蜍等动物的毒液,如: 敬钊缨毛蛛毒素-I[4]、敬钊缨毛蛛毒素-III[5]和GxTX-1E[6]等,但是这些多肽来源有限,限制了它们在药理学中的应用。目前文献报道的Kv2.1小分子阻断剂多数是通过普筛的方法发现的,抑制活性弱、选择性差,如: SC-791[1]、普罗帕酮[7]、三氟哌多[8]、17β-雌二醇[9]、布格呋喃 (AF-5)[10]等 (图 1)。2011年Merk公司研究人员[11]报道了通过高通量筛选获得了苯并咪唑类RY785和取代的苯甲酰胺类RY796 Kv2.1小分子阻断剂 (图 1),对Kv2.1具有较强的抑制活性和较好的选择性。因此,寻找新结构的高活性的Kv2.1小分子阻断剂具有重要意义[3, 11],它们不仅是研究Kv2.1生物学功能的分子探针,还有可能发展成为以Kv2.1为靶点的药物。本文依据文献报道的苯并咪唑类Kv2.1小分子阻断剂,设计合成了新结构的喹啉并吡咯烷和二氢喹啉并吡咯烷类衍生物并评价了合成化合物对Kv2.1的抑制活性,发现了两个新结构二氢喹啉类化合物 (3a和5a) 对Kv2.1具有抑制活性。
依据本课题组报道的合成方法[12],合成了关键中间体四氢喹啉并吡咯烷7。以吡咯烷-2-酮1为起始原料,经Cbz保护、DIBAL-H还原和消除反应,生成N-Cbz-2,3-二氢吡咯4。以六氟异丙醇为溶剂,对硝基苯胺5和乙醛酸乙酯6缩合生成的亚胺中间体作为双烯体,N-Cbz-2,3-二氢吡咯为亲双烯体,经aza-Diels- Alder反应,在一锅反应中缩合生成四氢喹啉并吡咯烷中间体7。化合物7为一对非对映异构体,其相对构型确证已经在文献中报道[12]。本文以非对映异构体的混合物7作为合成中间体,直接进行下一步反应,合成二氢喹啉并吡咯烷和喹啉并吡咯烷类衍生物 (合成路线1)。
从6-硝基四氢喹啉并吡咯烷7出发,在锌粉和盐酸的条件下,未得到预期的6-氨基四氢喹啉并吡咯烷,而是生成6-氨基-二氢喹啉并吡咯烷8,推测硝基还原为氨基,然后发生了氧化脱氢反应。化合物8与羧酸衍生物 (R1~R5) 在HATU的条件下缩合,生成二氢喹啉并吡咯烷类目标化合物1a~5a (合成路线1)。
四氢喹啉并吡咯烷化合物7在DDQ的作用下发生脱氢芳构化反应,生成喹啉并吡咯烷中间体9。在SnCl2的作用下,硝基还原为氨基,再与羧酸衍生物 (R1~R5) 在HATU的条件下缩合,生成目标化合物1b~5b; 然后催化氢化脱除Cbz,得到目标化合物1c~3c和5c (合成路线1)。
结果与讨论 1 化合物合成本文设计的目标化合物 (1a~5a、1b~5b、1c~3c和5c) 均未见文献报道,其结构经1H NMR、HR-MS (ESI) 确证,产物收率、理化常数及波谱数据见表 1,化合物的结构信息见表 2。
值得一提的是,6-氨基二氢喹啉并吡咯烷8的结构确证。6-硝基四氢喹啉并吡咯烷7在锌粉和盐酸条件下,可能生成1,4-二氢喹啉或1,2-二氢喹啉衍生物。通过质谱和氢谱推测生成了6-氨基二氢喹啉并吡咯烷,但化合物中双键的位置不确定。由于6-氨基二氢喹啉并吡咯烷在空气中不稳定,本文将其与间甲氧基苯甲酸缩合制备衍生物,推测可生成1,4-二氢喹啉1a或1,2-二氢喹啉1a'。在1H-1H COSY谱图中 (图 2),存在H4-Ha (δ 5.21,δ 3.51) 相关,在HMBC谱图中 (图 3),存在Ca-H4 (δ 42.16,δ5.21) 的C-H相关,由此确认化合物结构为1,4-二氢喹啉衍生物1a。因此,化合物7在锌粉和盐酸条件下,发生了氧化脱氢反应,生成了6-氨基-1,4-二氢喹啉并吡咯烷化合物8。

|
Figure 1 Some Kv2.1 inhibitors |

|
Scheme 1 The synthetic routes of target compounds 1a-5a,1b-5b,1c-3c,5c and two intermediates R-4,R-5 |
|
|
Table 1 Physical and spectral data of target compounds. ①CDCl3; ②DMSO-d6 |
|
|
Table 2 Inhibitory activity against Kv2.1 of tested compounds at 10 μmol·L-1 concentration. The racemic RY796 was used as a reference molecule,which showed IC50 value of 0.48 μmol·L-1. The reported IC50 of (S)-RY796 was 0.25 μmol·L-1 [11] |

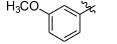
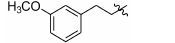
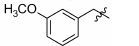

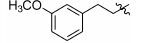


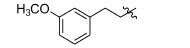
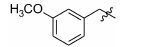
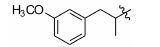
|
|
Figure 2 1H-1H COSY of compound 1a |

|
Figure 3 HMBC-NMR of compound 1a |
本文合成了二氢喹啉并吡咯烷和喹啉并吡咯烷类化合物14个,利用膜片钳技术测定了在10 μmol·L-1浓度下对Kv2.1的抑制活性 (表 2)。结果表明,化合物3a和5a对Kv2.1具有较好的抑制活性,抑制率分别为55.1% 和60.6%。因此,进一步测定了3a和5a的IC50值。如图 4所示,化合物3a和5a均能显著 抑制HEK293/Kv2.1钾电流,并且浓度依赖性地抑制Kv2.1活性,化合物3a和5a的IC50值分别为10.2和9.0 μmol·L-1。比较化合物3a (55.1%) 和3b(26.2%),5a (60.6%) 和5b (31.4%) 对Kv2.1的抑制率,可以看出,二氢喹啉类化合物具有较好的抑制活性。比较化合物1a~5a的抑制活性,发现在二氢喹啉的6-位引入间甲氧基苯丙酸和间甲氧基α-甲基苯丙酸时,化合物 (3a、5a) 具有显著的抑制活性,强于间甲氧基苯甲酸和间甲氧基苯乙酸取代的衍生物 (1a、2a),且强于肉桂酸衍生物 (4a),表明6-位取代基对活性具有显著性影响,苯丙酸类衍生物有利于提高化合物的抑制活性。

|
Figure 4 Effects of 5a and 3a on HEK293/Kv2.1. HEK293/ Kv2.1 was clamped at -70 mV and was depolarized from -50 mV to +50 mV for 300 ms in 20 mV increment,Kv2.1 current was measured at +50 mV. A and C: Typical recordings of 5a and 3a at 10 μmol·L-1 on Kv2.1. B and D: Dose- response curves of 5a and 3a on Kv2.1,the IC50 of 5a and 3a on Kv2.1 was 9.0 and 10.2 μmol·L-1,respectively |
本文从四氢喹啉并吡咯烷中间体出发,合成了新结构的二氢喹啉并吡咯烷和喹啉并吡咯烷类化合物,为二氢喹啉和喹啉衍生物的合成提供了一种新的合成方法。本文获得了新结构的Kv2.1抑制剂,二氢喹啉并吡咯烷类化合物3a和5a对Kv2.1抑制活性的IC50值分别为10.2和9.0 μmol·L-1。
实验部分熔点采用MP-J3型熔点仪,温度未校正; 1H NMR谱采用Varian 400 MHz Plus/BRUKER AV-III 500 MHz核磁共振仪测定 (TMS为内标); 质谱采用Thermo Exactive Plus质谱仪测定。所用试剂均为市售分析纯。
1 化学合成 1.1 N-Cbz-2-吡咯烷酮2的制备取2-吡咯烷酮 (1.7 g,20 mmol) 置于100 mL的圆底烧瓶中,加入25 mL无水THF,冷却至-40 ℃,加入NaH (880 mg,36.7 mmol) ,搅拌反应30 min。然后将氯甲酸苄酯 (3.74 g,22 mmol) 逐滴加入到上述反应体系中,加毕,继续于 -40 ℃反应,10 h后原料 消失,停止反应。加入50 ml水稀释反应液,用二氯甲烷萃取 (20 mL×3),二氯甲烷层依次用0.5 mol·L-1 HCl溶液 (20 ml×1)、饱和NaHCO3溶液 (20 ml×1) 和水 (20 ml×2) 洗,有机相用无水MgSO4干燥,浓缩得淡黄色油状物4.34 g,收率99.0%。1H NMR (400 MHz,CDCl3) δ: 7.47~7.29 (m,5H),5.28 (s,2H),3.82 (t,J = 8.0 Hz,2H),2.54 (t,J = 8.0 Hz,2H),2.07~2.00 (m,2H); ESI-MS m/z: 242 [M+Na]+。
1.2 N-Cbz-2-羟基吡咯烷3的制备取N-Cbz-2-吡咯烷酮 (4.29 g,19.60 mmol) 置 于100 mL的圆底烧瓶中,加入20 mL无水THF,冷却至 -78 ℃,Ar气保护下加入DIBAL-H甲苯溶液 (1.2 mol·L-1,31.2 mL),滴毕,于-78 ℃、Ar气保护下继续反应,5 h后原料反应完全,停止反应,加入饱和NH4Cl溶液 (20 mL),升至室温,加入饱和NaHCO3溶液 (30 mL) 后,二氯甲烷 (30 mL) 萃取,有机相用饱和NaCl (20 mL×2) 洗,无水MgSO4干燥,浓缩,得淡黄色油状物3.53 g,收率82.0%。1H NMR (400 MHz,CDCl3) δ: 7.37~7.31 (m,5H),5.54~5.49 (m,1H),5.18 (s,1H),5.15 (s,1H),3.39~3.32 (m,1H),3.66~3.56 (m,1H),2.09~1.83 (m,4H); ESI-MS m/z: 465 [2M+Na]+。
1.3 N-Cbz-2,3-二氢吡咯烷4的制备取N-Cbz-2-羟基吡咯烷 (3.53 g,15.97 mmol) 置于100 mL的圆底烧瓶中,加入30 mL甲苯和Ac2O (8.15 g,79.85 mmol)、DIEA (10.32 g,79.85 mmol),加热回流反应,6 h时后反应完全,浓缩,柱色谱 (乙酸乙酯-石油醚,体积比1∶30) 纯化,得到无色油状物1.63 g,收率50.3%。1H NMR (400 MHz,CDCl3) δ: 7.30~7.24 (m,5H),6.56 (brs,0.5H),6.47 (brs,0.5H),5.10 (s,2H),5.01 (brs,0.5H),4.96 (brs,0.5H),3.71 (dd,J = 18.8,9.2 Hz,2H),2.57 (dd,J= 17.6,9.6 Hz,2H); ESI-MS m/z: 204 [M+H]+。
1.4 化合物7的合成将对硝基苯胺 (576 mg,4.17 mmol) 置于50 mL圆底烧瓶中,加入六氟异丙醇 (15 mL),室温搅拌下加入乙醛酸乙酯的甲苯溶液 (852 mg,8.34 mmol),搅拌至反应完全; 然后加入N-Cbz-2,3-二氢吡咯烷,8 h后停止反应。浓缩,柱色谱分离 (乙酸乙酯-石油醚,体积比1∶2),得到两个化合物,分别为7-1和7-2,为一对非对映异构体。化合物7-1: 黄色固体,552 mg,收率31.5%; 1H NMR (400 MHz,CDCl3) δ: 8.53 (s,0.5H),8.37 (s,0.5H),7.94 (d,J = 8.4 Hz),7.52 (d,J = 6.8 Hz,1H),7.41~7.32 (m,4H),6.55 (d,J =9.2 Hz,1H),5.41(d,J = 7.2 Hz,0.5H),5.35 (d,J = 12.0 Hz,0.5H),5.29~5.23 (m,2H),5.14 (d,J = 8.8 Hz,1H),4.36~4.28 (m,3H),3.65 (dd,J = 19.2,9.2 Hz,0.5H),3.55 (dd,J= 19.2,9.6 Hz,0.5H),3.42~3.36 (m,1H),2.95 (m,1H),1.97~1.79 (m,2H),1.34 (t,J = 7.2 Hz,3H); ESI-MS m/z: 426 [M+H]+。化合物7-2,黄色固体,439 mg,收率25.1%; 1H NMR (400 MHz,CDCl3) δ: 8.52 (s,0.5H),8.38 (s,0.5H),7.95 (d,J = 8.8 Hz,1H),7.50~7.33 (m,5H),6.57 (d,J = 8.8 Hz,1H),5.34~5.03 (m,3H),4.91 (brs,1H),4.18 (q,J = 7.2 Hz,2H),4.04 (s,1H),3.57 (brs,1H),3.44 (brs,1H),2.96~2.90 (m,1H),2.18~2.12 (m,1H),2.01 (brs,1H),1.32 (t,J = 7.2 Hz,3H); ESI-MS m/z: 426 [M+H]+。
1.5 化合物8的合成将化合物7 (200 mg,0.47 mmol) 溶解于THF (10 mL) 中,加入Zn粉(130 mg,2.0 mmol),室温搅拌下加入2 mol·L-1的HCl溶液(1.5 mL),0.5 h后原料消失。向反应液中加入足量的饱和Na2CO3溶液,调pH为碱性。反应混合物中加入二氯甲烷 (15 mL×3) 萃取,有机相依次用水 (10 mL×1)、饱和氯化钠溶液 (10 mL×1) 洗,无水MgSO4干燥。浓缩,柱色谱分离 (乙酸乙酯-石油醚,体积比1∶2),得163 mg,黄色固体,收率88.0%; ESI-MS m/z: 394.23 [M+H]+。1H NMR (400 MHz,DMSO-d6) δ: 7.44~7.35 (m,5H),7.14 (d,J = 8.4 Hz,1H),6.62 (s,1H),6.48 (d,J = 7.7 Hz,1H),5.79 (s,1H),5.76 (s,1H),5.23 (brs,1H),5.15 (d,J = 9.2 Hz,2H),4.31~4.20 (m,1H),3.50 (brs,1H),3.32~ 3.21 (m,2H),2.20 (s,1H),1.58 (s,1H),1.28 (t,J = 7.1 Hz,1H); ESI-MS m/z: 394 [M+H]+。
1.6 化合物9的合成将化合物7 (200 mg,0.47 mmol) 溶解于DCM (10 mL) 中,加入DDQ (320 mg,1.41 mmol),回流 反应6 h,原料反应完毕。过滤,滤液用饱和Na2CO3 (10 mL×2)、蒸馏水 (10 mL×1)、饱和NaCl (10 mL×1) 洗涤,无水Na2SO4干燥,浓缩,重结晶 (乙酸乙酯-石油醚,体积比1∶2),得黄色固体183 mg,收率92.4%。1H NMR (400 MHz,CDCl3) δ: 9.35 (s,1H),8.40 (d,J = 9.3 Hz,1H),8.32 (d,J = 9.3 Hz,1H),7.44~7.33 (m,5H),5.36 (s,2H),4.54 (q,J = 7.2 Hz,2H),4.40 (t,J = 8.4 Hz,2H),3.63 (t,J = 8.4 Hz,2H),1.49 (t,J = 7.2 Hz,3H); ESI-MS m/z: 422 [M+H]+。
1.7 化合物10的合成将化合物9 (150 mg,0.36 mmol) 溶解于乙酸乙酯 (10 mL) 中,加入SnCl2·2H2O (403 mg,1.78 mmol),回流反应5 h后停止反应,加入5 mol·L-1 NaOH溶液(4 mL),过滤,依次用饱和NaHCO3溶液(10 mL×1)、水 (10 mL×2)、饱和NaCl溶液 (10 mL×1) 洗,无水Na2SO4干燥,浓缩,得黄色固体101 mg,72.6%。1H NMR (400 MHz,CDCl3) δ: 8.04 (d,J = 8.0 Hz,1H),7.45~7.35 (m,5H),7.10 (d,J = 8.0 Hz,1H),7.06 (s,1H),5.30 (s,2H),4.50 (q,J = 7.2 Hz,2H),4.30 (t,J = 8.0 Hz,2H),4.04 (brs,2H),3.53 (t,J = 8.0 Hz,2H),1.46 (t,J = 7.2 Hz,3H); ESI-MS m/z: 391 [M+H]+。
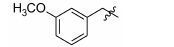
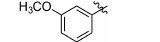
1.8 中间体R-4和R-5的制备 1.8.1 (E)-2-甲基-3-(3'-甲氧基苯基)丙烯酸 (R-4) 的制备 取间甲氧基苯甲醛 (1.0 g,7.35 mmol),依次加入丙酸钠 (776 mg,8.09 mmol) 和丙酸酐 (4.8 g,36.8 mmol),加热至160 ℃反应,17 h后停止反应。浓缩,倒入水中,过滤,滤饼溶于2 mol·L-1的NaOH溶液(7 mL) 中,用乙醚 (10 mL×4) 洗涤水层,合并醚层,用饱和NaHCO3溶液 (10 mL×4) 洗涤乙醚层,合并NaOH溶液和NaHCO3溶液,用盐酸酸化至pH 2~3,将析出的浅褐色固体过滤,再重结晶得白色固体877 mg,收率60.5%。1H NMR (400 MHz,CDCl3) δ:11.88 (s,1H),7.80 (s,1H),7.33 (t,J = 7.9 Hz,1H),7.03 (d,J = 7.6 Hz,1H),6.96 (s,1H),6.91 (d,J = 8.3 Hz,1H),3.84 (s,3H),2.15 (s,3H); ESI-MS m/z: 191 [M-H]-。
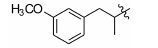
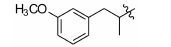
1.8.2 2-甲基-3-(3'-甲氧基苯基)丙酸 (R-5) 的制备 取2-甲基-3-(3'-甲氧基苯基) 丙烯酸 (R-4) (200 mg,1.04 mmol),加入20 mL无水甲醇,20 mg Pd/C,进行加压催化氢化 (压力: 50 psi),室温反应15 h。过滤,浓缩,得198 mg褐色油状物,收率98.0%。1H NMR (400 MHz,CDCl3) δ:7.26~7.18 (m,1H),6.79~6.74 (m,3H),3.79 (s,3H),3.06 (dd,J = 13.4,6.3 Hz,1H),2.81~2.73 (m,1H),2.64 (dd,J = 13.3,8.1 Hz,1H),1.18 (d,J = 6.9 Hz,3H); ESI-MS m/z: 193 [M-H]-。
1.9 化合物1a~5a、1b~5b、1c~3c和5c的合成 1.9.1 化合物1a~5a的合成(以化合物1a的合成为例) 取间甲氧基苯甲酸 (R-1) (79 mg,0.52 mmol),依次加入无水DCM (10 mL)、DIEA (100.6 mg,0.78 mmol) 和HATU (296 mg,0.78 mmol),搅拌30 min后,将化合物8(100 mg,0.25 mmol) 加入到上述体系中,室温反应8 h,原料反应完全。加入0.5 mol·L-1 HCl溶液(10 mL),依次用饱和碳酸钠 (10 mL)、蒸馏水 (10 mL)、饱和NaCl (10 mL) 洗,无水Na2SO4干燥,柱色谱 (乙酸乙酯-石油醚,体积比1∶2) 纯化,得白色固体57 mg,收率42.5%。化合物2a~5a的合成同1a。
1.9.2 化合物1b~5b的合成(以化合物1b的合成为例)取间甲氧基苯甲酸 (R-1) (79 mg,0.52 mmol),依次加入无水DMF (10 mL)、DIEA (100.6 mg,0.78 mmol) 和HATU (296 mg,0.78 mmol),搅拌30 min后,将化合物10 (100 mg,0.26 mmol) 加入到上述体系中,60 ℃反应8 h,原料反应完全。旋干后,加入20 mL乙酸乙酯,依次用饱和碳酸钠 (10 mL)、蒸馏水 (10 mL) 和饱和NaCl (10 mL) 洗,无水Na2SO4干燥,柱色谱 (乙酸乙酯-石油醚,体积比2∶3) 纯化,得微黄色固体48 mg,收率35.8%。化合物2b~5b的合成同1b。
1.9.3 化合物1c~3c和5c的合成(以化合物1c的合成为例) 取化合物1b (80 mg,0.15 mmol),加入乙醇 (10 mL) 和Pd/C (15 mg),常温常压下催化氢化,搅拌过夜,过滤,旋干溶剂,得黄色固体42 mg,收率71.2%。化合物2c、3c和5c的合成同1c。
2 Kv2.1抑制活性评价 2.1 细胞培养转染了人Kv2.1钾离子通道亚型的HEK293细胞 (HEK293/Kv2.1) 培养于含0.2 g·L-1 G418、10% 胎牛血清、10 000 u·L-1青霉素和链霉素的DMEM培养基中; 当细胞生长融合达到80% 时进行传代,用于后续电生理记录。
2.2 电生理实验记录Kv2.1钾通道电流的细胞外液成分 (mmol·L-1): NaCl 140、KCl 5、MgCl2 1、HEPES 10、glucose 10,pH 7.40; 电极内液成分 (mmol·L-1): KCl 140、MgCl2 2、EGTA 10、HEPES 10、ATP-2Na 2,pH 7.20。将培养的HEK293/Kv2.1用细胞外液漂洗2次后置于倒置相差显微镜下。玻璃微电极 (原料: GG17,外径: 15 mm,内径: 7.5 mm) 由P-97型拉制仪拉制,充灌电极内液后电阻为3~5 MΩ。在显微镜下选择边缘整齐、胞内无明显颗粒的细胞,移动电极并轻压细胞表面,用负压使电极尖端与细胞表面形成GΩ封接后,以较大负压破膜,对电容及电极串联阻抗进行补偿,形成全细胞记录方式。将细胞钳制在 -70 mV,从 -50 mV以20 mV的阶跃去极化到 +50 mV,持续300 ms,测量 +50 mV电位下的电流幅度即为Kv2.1钾电流。全细胞电流由EPC-10型膜片钳放大器记录,电流采用3 kHz低通滤波,采样率为20 kHz。应用Pulse 8.5软件进行刺激发放和信号采集。所有实验均在室温(23~25 ℃) 下进行。
| [1] | Frolov RV, Ignatova II, Singh S. Mechanism of K(v)2.1 channel inhibition by a selective COX-2 inhibitor SC-791 modification of gating[J]. Brain Res, 2010, 1359:67-74. |
| [2] | MacDonald PE, Sewing S, Wang J, et al. Inhibition of Kv2.1 voltage-dependent K+ channels in pancreatic β-cells enhances glucose-dependent insulin secretion[J]. J Biol Chem, 2002, 277:44938-44945. |
| [3] | Zhang HX. Role of Voltage-dependent Potassium Channels in Aβ25-35-induced Neuronal Apoptosis and the Relevant Pharmacological Study(电压依赖性钾通道在Aβ25-35 诱导的神经元凋亡中的作用及相关药理学研究)[D]. Beijing:Chinese Academy of Medicinal Sciences & Peking Union Medical College, 2004. |
| [4] | Tao H, Wu Y, Deng M, et al. Molecular determinants for the tarantula toxin jingzhaotoxin-I interacting with potassium channel Kv2.1[J]. Toxicon, 2013, 63:129-136. |
| [5] | Tao H, Chen JJ, Xiao YC, et al. Analysis of the interaction of tarantula toxin Jingzhaotoxin-Ⅲ (β-TRTX-Cj1α) with the voltage sensor of Kv2.1 uncovers the molecular basis for crossactivities on Kv2.1 and Nav1.5 channels[J]. Biochemistry, 2013, 52:7439-7448. |
| [6] | Lee S, Milescu M, Jung HH, et al. Solution structure of GxTX-1E, a high-affinity tarantula toxin interacting with voltage sensors in Kv2.1 potassium channels[J]. Biochemistry, 2010, 49:5134-5142. |
| [7] | Rolf S, Haverkamp W, Borggrefe M, et al. Effects of antiarrhythmic drugs on cloned cardiac voltage-gated potassium channels expressed in Xenopus oocytes[J]. Naunyn-Schmiedeberg's Arch Pharmacol, 2000, 362:22-31. |
| [8] | Wible B, Murawsky MK, Crumb WJ, et al. Stable expression and characterization of the human brain potassium channel Kv2.1:blockade by antipsychotic agents[J]. Brain Res, 1997, 761:42-50. |
| [9] | Zhang W, Jin HW, Zhang HX, et al. Effect of 17β-estradiol on Kv2.1 current and delayed rectifier potassium current cultured rat hippocampal neurons[J]. Acta Pharm Sin (药学学报), 2004, 39:686-690. |
| [10] | Chen CL, Wang WP, Wang L, et al. Electrophysiology mechanisms of 4-butyl-alpha-agarofuran:a new anxiolytic and antidepressant drug[J]. Acta Pharm Sin (药学学报), 2013, 48:38-44. |
| [11] | Herrington J, Solly K, Ratliff KS, et al. Identification of novel and selective Kv2 channel inhibitors[J]. Mol Pharmacol, 2011, 80:959-964. |
| [12] | Zhou J, Xu BL. Three-component one-pot synthesis of 1,2, 3,4-tetrahydroquinoline derivatives in hexafluoroisopropanol[J]. Chin Chem Lett, 2008, 19:921-924. |