 2016, Vol. 51
2016, Vol. 51



蛋白酶活化受体 (protease-activated receptors, PARs) 属于G蛋白偶联受体 (guanosine-binding protein coupled receptor, GPCR) 家族成员, 包含PAR1、PAR2、PAR3和PAR4。其中PAR2分布在许多组织和细胞中 (皮肤、呼吸道上皮细胞和胰腺等), 具有广泛的生物学效应, 其激活、灭活、脱敏、复敏及其与信号转导途径的关系, 尤其是参与很多疾病的发生和发展备受关注。本文就PAR2的结构与活化、相关靶点药物的研究进展综述如下。
1 PAR2结构与活化PAR2是由细胞外区 (N-末端和细胞外袢)、跨膜区 (7个跨膜螺旋) 及细胞内区 (细胞内袢和C-末端) 组成 (图 1)。蛋白酶 (如胰蛋白酶[1]、MMP-1[2, 3]) 通过裂解PAR胞外N-末端, 去除结合在受体表面的绳系配体, 从而激活跨膜信号传递至胞内G蛋白。根据已有的关于PAR2活化后的信号转导研究结论, 其细胞内的信号转导主要路径为PLC-Ca2+-PKC-MAPK路径以及MAPK-JNK-c-Jun、MAPK-JNK-P53和MAPK-JNK-STAT3等路径[4]。
 | Figure 1 The structure of protease-activated receptor 2 (PAR2) |
PAR2连接配体的合成是根据天然系配体结构-SLIGKV (人) 和SLIGRL (啮齿类动物) 设计的[5]。研究[6]报道, 将SLIGRL-NH2中的丝氨酸(serine) 替换成2-furoyl (2f) 基团并且在C端加入一个鸟氨酸 (ornithine), 合成为2f-LIGRLO-NH2, 可以显著提高至少10倍的PAR2激动剂活性, 而且 对PAR2的结合选择性高于PAR1, 在细胞中检测出其动员iCa2+ 释放EC50为0.2 μmol·L-1。也有研究[7]报道在SLIGRL-NH2 (化合物1) (表 1) 的C端增加 一个第七基团, 如异亮氨酸 (isoleucine), 可以提高 6倍激动活性; 另外将第六基团亮氨酸 (leucine) 替换为芳香基团, 如酪氨酸 (tyrosine)、4-硝基苯丙氨 酸 (4-nitrophenylalanine) 或3,4-二氯苯丙氨酸 (3,4- dichlorophenylalanine), 都可以增加激动活性。理论 上认为用4-硝基苯丙氨酸 (4-nitrophenylalanine) 取代2f-LIGRLI-NH2 (化合物2) 的第六基团也可能提高 激动活性, 但结果却没观察到激动活性有所提高, 这个肽和2f-LIGRLI-NH2在HT29细胞中动员iCa2+释放有相同的活性 (EC50 = 0.2 μmol·L-1)。该报道[7]中 还研究了用一系列的杂环和芳香基团如4-(2-methyloxazolyl)、5-isoxazolyl、2-pyrazoyl、2-pyridoyl、3- pyridoyl、2-benzofuranoyl、2-naphthoyl和2-benzothienyl替代SLIGRLI-NH2 (化合物3) 中的丝氨酸基团所有合成出来的产物 (化合物4) 的激动活性EC50为0.1~0.3 μmol·L-1 (iCa2+、HT29细胞)。
|
|
Table 1 PAR2-target medicines |

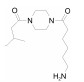







SLIGRL-NH2的丝氨酸与取代物基团 [杂环基团、2-aminothiazol-4-oyl (2-at) 和6-aminonicotinoyl (6-an)] 类似, 都可以合成出2f-LIGRLO-NH2的激动剂类似物, 同时激活Ca2+ 和MAPK信号转导[8]。在2-atLIGRLO-NH2的鸟氨酸侧链用3个聚乙二醇 (polyethylene glycol, PEG) 连接一个棕榈酰基团可以使PAR2增加200倍的激动活性 (EC50 = 4 nmol·L-1, iCa2+; EC50 = 10 nmol·L-1, MAPK; 16HBE细胞)[9]。通过研究不同链长的疏水性链接物, PEG3-Pam是最佳的链接物。研究报道, 在Ca2+脱敏实验中2-atLIGRLO (PEG3-Pam)-NH2 (化合物5) 对PAR2的结合选择性高于PAR1。
通过在化合物库中筛选得到小分子PAR2激动剂AC-55541 (化合物6)、AC-98170 (化合物7)、AC- 264613 (化合物8)[10, 11], 采用人HEK293T细胞, 进行细胞增殖、磷脂酰肌醇水解和动员Ca2+释放实验。结果显示, AC-55541和AC-264613是PAR2完全激动 剂(EC50分别为200~1000和30~100 nmol·L-1), 而AC-98170是PAR2部分激动剂。当AC-264613在HT29细胞中实验时, 未发现其激动剂活性, 并且浓度为1 μmol·L-1时也不刺激Ca2+释放, 所以AC-264613在不同细胞种类和条件下是否具有介导PAR2活性仍然需要继续考察。
多肽受体激动剂的脂化通过特有的机制可以用于提高效能[12]。2at-LIGRL-PEG3-Hdc (化合物9) 通过3个PEG连接一个十六烷基 (hexadecyl, Hdc) 脂质, 具有强有力的PAR2激动剂活性 (EC50 = 1.4 nmol·L-1)[13]。在一系列截短类似物中, 2at-LIGR- PEG3-Hdc (化合物10) 仍然保持着PAR2激动剂活 性 (EC50 = 2.1 nmol·L-1), 且对PAR2具有高度选择 性; 2at-LIG-PEG3-Hdc是最短的PAR2完全激动剂, 尽管其EC50增加为46 nmol·L-1, 2at-LI-PEG3-Hdc 仍然保留对PAR2特异激活性, 但是在生理活性和Ca2+信号中只能部分激活PAR2; 而2at-L-PEG3-Hdc和2at-PEG3-Hdc这两个截短类似物在体外实验中并未观察到具有激活PAR2的活性[13]。GB110 (化合物11)[14]是一个强有力且具有选择性的激动剂 (EC50 = 0.28 μmol·L-1, iCa2+, HT29细胞), 这是迄今为止报道的最有力的PAR2肽类似物激动剂。
2.2 PAR2拮抗剂FSLLRY-NH2 (化合物12) 和LSIGRL-NH2 (化合物13) 是PAR2多肽拮抗剂, 可 以抑制胰蛋白酶诱导的PAR2活性 (IC50 = 50~200 μmol·L-1, iCa2+, PAR2转染KNRK细胞), 而对SLIGRL- NH2诱导的PAR2活性没作用[15]。首次报道[16]的非肽PAR2拮抗剂是ENMD-1068 (化合物14), 体外实验中其对PAR2的特异性高于其他PARs, 但效能较低 (IC50 = 5 mmol·L-1)。
2009年, 研究[17]报道K-14585 (化合物15) 是一个效能更强的PAR2拮抗剂。K-14585不能抑制蛋白酶 (如胰蛋白酶) 诱导的PAR2活性, 但是它可以竞争性拮抗3H-2f-LIGRL-NH2结合到NCTC2544-PAR2细胞上 (Ki = 0.6 μmol·L-1)。K-14585在某些信号传导中起双重作用, 如PAR2介导的ERK磷酰化、p38 MAPK和NF-κB通路, 在低浓度时可以抑制SLIGKV-NH2诱导的信号传导, 但在高浓度时起激动剂作用[18]。由于不具备抑制内源性蛋白酶诱导的PAR2活化, K-14585的应用有所限制。
化合物16(IC50 = 57 μmol·L-1, iCa2+, HT29细 胞)、GB83 (化合物17, IC50 = 2 μmol·L-1) 和GB88 (化合物18, IC50 = 1 μmol·L-1) 都是从非肽PAR2激 动剂GB110衍生的类似物[14, 19]。其中, GB88是第一个可以抑制所有已知PAR2激动剂 (包括合成肽如SLIGKV-NH2、SLIGRL-NH2和2f-LIGRLONH2; 非肽激动剂GB110; 内源性激动剂如胰蛋白酶和类胰蛋白酶) 诱导的iCa2+释放的非肽PAR2拮抗剂。这使得GB88在动物疾病模型中研究拮抗PAR2的作用中发挥极大的作用, 包括大鼠足肿胀、结肠炎、关节炎和食源性代谢障碍等模型。口服GB88 (5~10 mg·kg-1) 可以缓解PAR2激动剂诱导的足肿胀和急性炎症[19], 同样也可以抑制大鼠胶原诱导性关节炎[20]。大鼠每天口服GB88 (10 mg·kg-1) 可以抑制PAR2激动剂诱导的急性结肠炎和TNBS诱导的慢性结肠炎[21]。如果每天口服GB88 (10 mg·kg-1), 可以缓解大鼠饮食诱导性肥胖, 抑制心血管重构和心肌纤维化、胰岛素敏感性和葡萄糖耐受不良、肝脏和胰腺功能紊乱, 这说明抑制PAR2可以有效治疗和预防新陈代谢功能紊乱[22]。GB88是目前用于体内抗炎的最有效的小分子PAR2拮抗剂。这些拮抗剂在体外信号机制和体内PAR2介导的不同疾病模型研究中提供了很有效的帮助。
2.3 PepducinCovic等[23]首次报道了pepducin技术, 这是一个全新的方法: pepducin由一个脂质部 分 (如棕榈酸酯、肉豆蔻酸酯或石胆酸), 附着于一 个靶向于GPCR的胞质循环 (C1、C2或C3) 或C端末尾 (C4) 上的多肽组成, 通过GPCRs与细胞膜上的G-proteins相互作用来调节GPCR的活性。由于多肽结构的不同, pepducin可以表现为GPCR的激动剂、拮抗剂及活性调节剂。
P2pal-21是PAR2部分激动剂, 在PLC-β/InsP通路中表现为浓度依赖性双重活性, 浓度小于100 nmol·L-1时为部分激动剂, 高浓度 (IC50=1 μmol·L-1)时可以阻断胞外激活剂SLIGKV-NH2 (100 μmol·L-1)引起的PAR2信号通路[23]。P2pal-21上的单个残基K287F突变得到P2pal-21F, 这是一个PAR2完全激动剂, 同时可以激活PAR1。
Sevigny 等[24]采用了一个PAR2同源二聚体的分子模型和第三胞内环 (C3)的突变分析的方法, 明确了控制固有活性的关键基团。通过这个结构设计出P2pal-18S, 体外实验表明, 它可以完全抑制PAR2介导的信号通路, 并且在小鼠模型上有效阻断PAR2依赖性炎症反应。
3 结论与展望PAR2在人体内各组织和细胞中广泛分布, 在某些病理状态下表达显著上调或下降, 是某些疾病发病机制的一个重要调节因子, 可能放大炎症反应和感染或物理损伤中的其他信号反应。因此, 有效的、选择性的PAR2激动剂或拮抗剂不仅可以更好地研究PAR2在炎症、呼吸疾病、新陈代谢和心血管疾病等病理和生理功能及其作用机制, 也可以评估PAR2作为疾病治疗靶点的可行性。
目前, 以PAR2为治疗靶点的研究主要集中在直接干预PAR2激活, 包括抑制胰蛋白酶活性、单克隆抗体封闭PAR2受体和阻断PAR2活化的小分子化 合物等方面。由于激活PAR2的物质并不单一, PAR2分布的广泛性导致单克隆抗体潜在的不良反应, 此方面的研究离实际应用还有一定距离。而pepducin可以快速传导至细胞质膜并在膜周围靶标达到较高有效浓度, 具有超越膜障碍, 以及细胞内受体结构的丰富多样性可能成为新的治疗药物及用于研究G蛋白偶联受体作用机制。因此, pepducin可能会在以PAR2为靶点的研究上有重大突破, 成为新的实验途径突破点。
| [1] | Nystedt S, Emilsson K, Wahlestedt C, et al. Molecular cloning of a potential proteinase activated receptor[J]. Proc Natl Acad Sci USA, 1994, 91:9208-9212. |
| [2] | Boire A, Covic L, Agarwal A, et al. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells[J]. Cell, 2005, 120:303-313. |
| [3] | Trivedi V, Boire A, Tchernychev B, et al. Platelet matrix metalloprotease-1 mediates thrombogenesis by activating PAR1 at a cryptic ligand site[J]. Cell, 2009, 137:332-343. |
| [4] | Ramachandran R, Eissa A, Mihara K, et al. Proteinaseactivated receptors (PARs):differential signalling by kallikreinrelated peptidases KLK8 and KLK14[J]. Biol Chem, 2012, 393:421-427. |
| [5] | Macfarlane SR, Seatter MJ, Kanke T, et al. Proteinaseactivated receptors[J]. Pharmacol Rev, 2001, 53:245-282. |
| [6] | Mcguire JJ, Saifeddine M, Triggle CR, et al. 2-Furoyl-LIGRLO-amide:a potent and selective proteinase-activated receptor 2 agonist[J]. J Pharmacol Exp Ther, 2004, 309:1124-1131. |
| [7] | Barry GD, Suen JY, Low HB, et al. A refined agonist pharmacophore for protease activated receptor 2[J]. Bioorg Med Chem Lett, 2007, 17:5552-5557. |
| [8] | Boitano S, Flynn AN, Schulz SM, et al. Potent agonists of the protease activated receptor 2(PAR2)[J]. J Med Chem, 2011, 54:1308-1313. |
| [9] | Flynn AN, Hoffman J, Tillu DV, et al. Development of highly potent protease-activated receptor 2 agonists via synthetic lipid tethering[J]. FASEB J, 2013, 27:1498-1510. |
| [10] | Gardell LR, Ma JN, Seitzberg JG, et al. Identification and characterization of novel small-molecule protease-activated receptor 2 agonists[J]. J Pharmacol Exp Ther, 2008, 327:799-808. |
| [11] | Seitzberg JG, Knapp AE, Lund BW, et al. Discovery of potent and selective small-molecule PAR-2 agonists[J]. J Med Chem, 2008, 51:5490-5493. |
| [12] | Zhang L, Bulaj G. Converting peptides into drug leads by lipidation[J]. Curr Med Chem, 2012, 19:1602-1618. |
| [13] | Boitano S, Hoffman J, Tillu DV, et al. Development and evaluation of small peptidomimetic ligands to proteaseactivated receptor-2(PAR2) through the use of lipid tethering[J]. PLoS One, 2014, 9:e99140. |
| [14] | Barry GD, Suen JY, Le GT, et al. Novel agonists and antagonists for human protease activated receptor 2[J]. J Med Chem, 2010, 53:7428-7440. |
| [15] | Al-Ani B, Saifeddine M, Wijesuriya SJ, et al. Modified proteinase-activated receptor-1 and -2 derived peptides inhibit proteinase-activated receptor-2 activation by trypsin[J]. J Pharmacol Exp Ther, 2002, 300:702-708. |
| [16] | Kelso EB, Lockhart JC, Hembrough T, et al. Therapeutic promise of proteinase-activated receptor-2 antagonism in joint inflammation[J]. J Pharmacol Exp Ther, 2006, 316:1017-1024. |
| [17] | Kanke T, Kabeya M, Kubo S, et al. Novel antagonists for proteinase-activated receptor 2:inhibition of cellular and vascular responses in vitro and in vivo[J]. Br J Pharmacol, 2009, 158:361-371. |
| [18] | Goh FG, Ng PY, Nilsson M, et al. Dual effect of the novel peptide antagonist K-14585 on proteinase-activated receptor-2 mediated signaling[J]. Br J Pharmacol, 2009, 158:1695-1704. |
| [19] | Suen JY, Barry GD, Lohman RJ, et al. Modulating human proteinase activated receptor 2 with a novel antagonist (GB88) and agonist (GB110)[J]. Br J Pharmacol, 2012, 165:1413-1423. |
| [20] | Lohman RJ, Cotterell AJ, Barry GD, et al. An antagonist of human protease activated receptor-2 attenuates PAR2 signaling, macrophage activation, mast cell degranulation, and collageninduced arthritis in rats[J]. FASEB J, 2012, 26:2877-2887. |
| [21] | Lohman RJ, Cotterell AJ, Suen J, et al. Antagonism of protease-activated receptor 2 protects against experimental colitis[J]. J Pharmacol Exp Ther, 2012, 340:256-265. |
| [22] | Lim J, Iyer A, Liu L, et al. Diet-induced obesity, adipose inflammation, and metabolic dysfunction correlating with PAR2 expression are attenuated by PAR2 antagonism[J]. FASEB J, 2013, 27:4757-4767. |
| [23] | Covic L, Gresser AL, Talavera J, et al. Activation and inhibition of G protein-coupled receptors by cell-penetrating membrane-tethered peptides[J]. Proc Natl Acad Sci USA, 2002, 99:643-648. |
| [24] | Sevigny LM, Zhang P, Bohm A, et al. Interdicting proteaseactivated receptor-2-driven inflammation with cell-penetrating pepducins[J]. Proc Natl Acad Sci USA, 2011, 108:8491-8496. |